Оригинальные и воспроизведенные лекарственные препараты: что нужно знать клиницисту?
СтатьиА.В. Щулькин, А.А. Филимонова
ФГБОУ ВО «Рязанский государственный медицинский университет» Минздрава России, Рязань, Россия
Резюме
В обзорной статье рассмотрены современные подходы к тестированию и регистрации воспроизведенных (генерических) лекарственных препаратов. Представлена история формирования методологии исследований воспроизведенных лекарственных препаратов и современное законодательство Российской Федерации. Описаны этапы подтверждения эквивалентности оригинального и воспроизведенных лекарственных препаратов: фармацевтическая эквивалентность, биоэквивалентность и терапевтическая эквивалентность. Подробно разобраны способы оценки биоэквивалентности как основного исследования при регистрации воспроизведенных лекарственных препаратов. На примере оригинального отечественного нейропротектора препарата Мексидол (этилметилгидроксипиридина сукцината) и его генериков описано, как реализуются законодательные акты на практике. Делается вывод о том, что не все воспроизведенные лекарственные препараты могут быть взаимозаменяемыми с оригинальным препаратом.
Ключевые слова: оригинальный и воспроизведенные препараты, генерики, этилметилгидроксипиридина сукцинат, Мексидол.
Информация об авторах:
Щулькин А.В. — orcid.org/0000-0003-1688-0017
Филимонова А.А. — orcid.org/0000-0001-7524-3195
Автор, ответственный за переписку: Щулькин А.В. — e-mail: alekseyshulkin@rambler.ru
Как цитировать: Щулькин А.В., Филимонова А.А. Оригинальные и воспроизведенные лекарственные препараты: что нужно знать клиницисту? Журнал неврологии и психиатрии им. С.С. Корсакова. 2021;121(10):99—104. doi.org/10.17116/jnevro202112110199
Лекарственная терапия является одним из основных методов лечения в XXI веке. В 2016 г. США потратили 450 млрд долларов на рецептурные препараты, что составило 14% от общих затрат на здравоохранение. По прогнозам, данная сумма составит 610 млрд долларов в 2021 г. [1]. Большая часть этих расходов (более 70%) приходится на оригинальные лекарственные препараты, хотя они составляют только 10% рецептов, отпускаемых в США [2].
Одним из подходов к снижению затрат на здравоохранение является дальнейшее расширение использования воспроизведенных (генерических) лекарственных препаратов [2]. Принципиально важно, чтобы уменьшение стоимости лечения не сопровождалось снижением его качества. На достижение оптимального соотношения цена—качество и нацелено постоянное совершенствование требований к регистрации воспроизведенных лекарственных препаратов.
В законодательстве РФ за последнее десятилетие также произошли изменения в регистрации и использовании генериков, которые не всегда хорошо известны клиницистам, освещению которых посвящен настоящий обзор.
История развития подходов к тестированию воспроизведенных лекарственных препаратов
Основоположником подходов к тестированию воспроизведенных лекарственных препаратов и разработчиком современных общепризнанных рекомендаций является FDA (США), поэтому историю развития биоэквивалентности в этой стране можно рассматривать как мировую [3]. В начале 1970-х годов в США было выявлено, что пациенты, принимавшие дигоксин, имеют вариабельный или слабый ответ на проводимую терапию. При изучении механизмов развития такого феномена J. Lindenbaum и соавт. в перекрестном исследовании на четырех добровольцах показали, что после перорального приема дигоксина разных производителей в дозе 0,5 мг наблюдались выраженные различия максимальной концентрации вещества (Cmax) в сыворотке крови [4]. Один препарат показал в 7 раз более высокие концентрации дигоксина, чем рецептура другого производителя. Значительные различия наблюдались даже между разными партиями одного и того же препарата. Данные результаты были подтверждены и в других исследованиях [5, 6]. Вероятными причинами различий были недостаточное или чрезмерное количество действующего вещества в принимаемых препаратах, разные размер частиц, время распада, скорость растворения и влияние наполнителей. Было установлено, что значительные колебания концентраций лекарственных веществ в плазме крови могут подвергать пациентов потенциально смертельной опасности, поэтому группа экспертов FDA в 1977 г. разработала первые рекомендации по исследованию генериков [3], которые в дальнейшем неоднократно обновлялись и пересматривались.
Оригинальные и воспроизведенные лекарственные препараты в современном законодательстве РФ
Согласно Федеральному закону от 27 декабря 2019 г. №475-ФЗ «О внесении изменений в Федеральный закон «Об обращении лекарственных средств», под термином «оригинальный лекарственный препарат» подразумевается препарат с новым действующим веществом, который первым зарегистрирован в РФ или в иностранных государствах на основании результатов доклинических исследований лекарственных средств и клинических исследований лекарственных препаратов, подтверждающих его качество, эффективность и безопасность [7].
Термин «воспроизведенный лекарственный препарат» подразумевает лекарственный препарат для медицинского применения, который имеет эквивалентный референтному лекарственному препарату качественный и количественный состав действующих веществ в эквивалентной лекарственной форме, биоэквивалентность или терапевтическая эквивалентность которого соответствующему референтному лекарственному препарату подтверждена соответствующими исследованиями [7].
Биоэквивалентность лекарственных препаратов — достижение сопоставимых показателей скорости всасывания, степени поступления к месту действия и скорости выведения одного или нескольких обладающих фармакологической активностью действующих веществ при назначении лекарственных препаратов для медицинского применения, имеющих одно международное непатентованное (химическое, группировочное) наименование, в эквивалентных дозировках и при одинаковом способе введения [7].
Терапевтическая эквивалентность лекарственных препаратов — достижение клинически сопоставимых терапевтического эффекта и показателей эффективности и безопасности при назначении лекарственных препаратов для медицинского применения, имеющих одно международное непатентованное (химическое, группировочное) наименование, в эквивалентных дозировках по одним и тем же показаниям к применению и при одинаковом способе введения у одной и той же группы больных [7].
Дополнительно выделено понятие «референтный лекарственный препарат», под которым понимают лекарственный препарат, используемый для оценки биоэквивалентности или терапевтической эквивалентности, качества, эффективности и безопасности воспроизведенного лекарственного препарата [7].
Подчас у практикующих врачей трудности возникают с интерпретацией понятия «биоэквивалентность», поэтому остановимся на нем подробнее. В основе классической фармакологии лежит представление о том, что лекарственный препарат оказывает свое действие за счет воздействия на специфические мишени. Поэтому принято считать, что для проявления одинакового фармакологического эффекта оригинальный и воспроизведенный лекарственные препараты должны обеспечивать одинаковую концентрацию действующего вещества в области своей мишени (так как определение концентрации веществ в области мишени практически невозможно, то на практике используют определение концентрации в плазме крови, при этом предполагая, что концентрация в плазме крови и в области мишени находятся в динамическом равновесии) [8].
Различия между оригинальным и воспроизведенным лекарственным препаратом проявляются только на этапе их абсорбции (всасывания). Остальные этапы фармакокинетики (биотрансформация, выведение и распределение) у оригинального и воспроизведенного препаратов эквивалентные, так как они содержат одно и тоже действующее вещество в одной и той же дозе. Поэтому при регистрации водных инъекционных лекарственных препаратов исследования по оценке биоэквивалентности, как правило, не проводят (биодоступность при внутривенном введении составляет 100%) [9]. Иными словами, в ходе выполнения исследований биоэквивалентности лекарственных препаратов оценивают их относительную биодоступность.
 Биодоступность (биологическая доступность) — скорость и степень, с которой действующее вещество абсорбируется из лекарственного препарата и становится доступным в месте своего действия. Относительную биодоступность действующего вещества в определенной лекарственной форме определяют путем сравнения с биодоступностью другой лекарственной формы, введенной тем же или другим (но не внутривенным) путем [9].
Биодоступность (биологическая доступность) — скорость и степень, с которой действующее вещество абсорбируется из лекарственного препарата и становится доступным в месте своего действия. Относительную биодоступность действующего вещества в определенной лекарственной форме определяют путем сравнения с биодоступностью другой лекарственной формы, введенной тем же или другим (но не внутривенным) путем [9].
Согласно современным рекомендациям, оценка биоэквивалентности может выполняться в исследованиях in vivo и in vitro [9, 10, 11]:
— сравнительные фармакокинетические исследования;
— сравнительные фармакодинамические исследования;
— сравнительные клинические испытания;
— сравнительные тесты in vitro.
Наиболее точным, чувствительным и воспроизводимым методом является исследование биоэквивалентности in vivo с оценкой относительной биодоступности оригинального и воспроизведенного лекарственных препаратов, то есть фармакокинетическое исследование [3].
Эти исследования обычно проводятся с ограниченным числом здоровых добровольцев, например, с 24—36 субъектами. Большинство исследований имеют двухпоследовательный, двухпериодный, перекрестный дизайн, в которых каждому из участников случайным образом назначается последовательность: воспроизведенный (тест) препарат — оригинальный (референс) препарат или оригинальный (референс) препарат — воспроизведенный (тест) препарат с адекватным интервалом вымывания между двумя периодами лечения [11].
После приема тестируемых препаратов у добровольцев оценивают концентрацию действующего вещества в плазме крови и строят фармакокинетическую кривую — график зависимости изменения концентрации действующего вещества от времени. Скорость абсорбции вещества оценивают с помощью таких показателей, как Cmax и время достижения максимальной концентрации (Tmax). Степень абсорбции характеризуют по площади под фармакокинетической кривой концентрация—время (англ.: area under curve AUC). AUC показывает общее количество лекарственного вещества, поступившего в системный кровоток (см. рисунок) [12].
Фармакокинетическая кривая зависимости концентрации вещества в плазме крови от времени. (Pharmacokinetic curve of the dependence of the substance concentration in the blood plasma from time.)
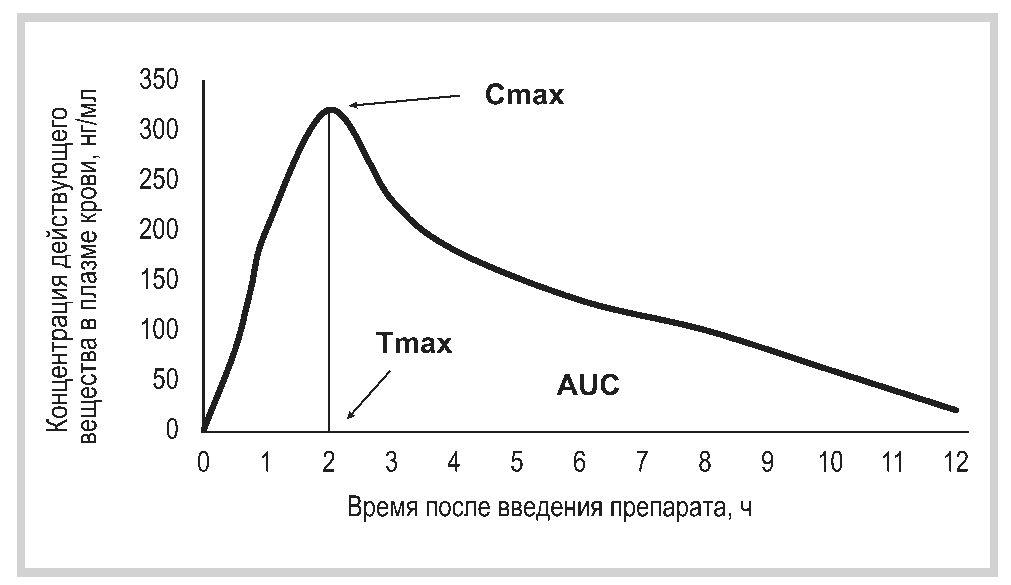
Эксперты FDA предложили считать значимой разницу между значениями Cmax, AUC после приема оригинального и воспроизведенного лекарственных препаратов более 20%, поскольку изменение этих показателей более 20% может сопровождаться развитием токсических эффектов проводимой терапии. При этом важна не точечная оценка (сравнение средних значений), а определение доверительного интервала изменений данных фармакокинетических параметров: рассчитывают 90% доверительный интервал отношения средних геометрических параметров, который должен укладываться в интервал 80—125%. Снижение Cmax и AUC воспроизведенного лекарственного препарата по сравнению с оригинальным ниже 80% может приводить к уменьшению эффективности проводимой терапии, а превышение 125% — к повышению риска развития токсических эффектов [3].
Одним из потенциальных недостатков такого подхода является то, что он не может ответить на вопрос о том, достаточно ли установление биоэквивалентности для того, чтобы гарантировать, что отдельный пациент может одинаково реагировать на два разных продукта. Это связано с тем, что используемый подход оценивает только разницу между средними значениями показателей Cmax и AUC оригинального и воспроизведенного лекарственных препаратов, в то время как индивидуальная биоэквивалентность характеризует разницу между средним значением и вариабельностью. Anderson S. и соавт. предложили подход для оценки индивидуальной биоэквивалентности, чтобы гарантировать, возможность перевода отдельного пациента с оригинального лекарственного препарата на генерик [13, 14]. Однако данный подход пока не внедрен в клиническую практику.
В 1995 г. G. Amidon и соавт. разработали Биофармацев-тическую классификационную систему (БКС) для оценки зависимости растворения лекарственного препарата in vitro и биодоступности in vivo [15]. Эта классификация была основана на физических свойствах растворимости и проницаемости лекарственного средства при абсорбции. Согласно БКС, лекарственные вещества подразделяются на четыре класса (см. таблицу). Если лекарственное средство в максимальной концентрации в тестируемой лекарственной форме растворимо в 250 мл или менее водной среды в диапазоне pH 1,0—7,5, то лекарственный препарат считается хорошо растворимым. Лекарственный препарат считается высокопроницаемым, если определено, что степень его кишечного всасывания составляет 90% или выше.
Таблица. Биофармацевтическая классификационная система/ Biopharmaceutical Classification System
|
Биофармацевтический класс |
Растворимость |
Проницаемость |
|
I |
Высокая |
Высокая |
|
II |
Низкая |
Высокая |
|
III |
Высокая |
Низкая |
|
IV |
Низкая |
Низкая |
Согласно современным подходам, для лекарственных препаратов немедленного высвобождения, хорошо растворимых и подвергающихся полной абсорбции (I класс по БКС), а также для лекарственных препаратов хорошо растворимых, но подвергающихся ограниченной абсорбции (III класс по БКС) допускается регистрация без проведения исследований in vivo, то есть без оценки относительной биодоступности на здоровых добровольцах [9]. Процедура оценки биоэквивалентности лекарственного препарата без проведения исследования in vivo получила название «биовейвер» (от англ.: biowaiver). Другими словами, регистрация воспроизведенных лекарственных препаратов с немедленным высвобождением возможна и на основе только данных in vitro. Этот подход основывается на том, что если препарат быстро растворяется в желудочнокишечном тракте, то его абсорбция существенно не зависит от других факторов [3].
Если в ряде случаев фармакокинетический подход в оценке биоэквивалентности невозможен, тогда выполняют фармакодинамические и клинические исследования (часто путают с оценкой терапевтической эквивалентности, хотя это разные исследования).
Исследование фармакодинамической эквивалентности может понадобиться, когда количественное определение содержания действующего вещества и/или его метаболитов в плазме крови или моче не может быть проведено с достаточной чувствительностью и точностью. Кроме того, фармакодинамические исследования эквивалентности у человека необходимы, если измерение концентраций действующего вещества не может быть использовано в качестве суррогатных конечных точек для подтверждения эффективности и безопасности конкретного лекарственного препарата, например, оказывающего местное действие [9, 10].
Несмотря на то что фармакодинамические исследования могут быть подходящим инструментом для установления эквивалентности, иногда они не могут быть использованы из-за отсутствия значимых фармакодинамических параметров, которые могут быть достоверно измерены. В этом случае для подтверждения эквивалентности двух лекарственных препаратов необходимо проведение клинических исследований. Необходимо подчеркнуть, что оценивать эквивалентность предпочтительнее, проводя фармакокинетические исследования вместо клинических, так как последние обладают меньшей чувствительностью и могут потребовать включения значительного количества субъектов для достижения достаточной статистической мощности [9, 10].
Понятие о взаимозаменяемых лекарственных препаратах
Федеральным законом РФ №429 от 22 декабря 2014 г. «О внесении изменений в Федеральный закон "Об обращении лекарственных средств"» было введено понятие «взаимозаменяемый лекарственный препарат» [16]. Так, взаимозаменяемый лекарственный препарат — лекарственный препарат с доказанной терапевтической эквивалентностью или биоэквивалентностью в отношении референтного лекарственного препарата, имеющий эквивалентные ему качественный и количественный состав действующих веществ, состав вспомогательных веществ, лекарственную форму и способ введения.
Комиссия экспертов экспертного учреждения дает заключение о взаимозаменяемости лекарственного препарата на основании следующих критериев (характеристик): фармацевтической эквивалентности, биоэквивалентности и/ или терапевтической эквивалентности; идентичности способа введения и способа применения; соответствия производителя лекарственного средства требованиям правил надлежащей производственной практики.
Важно отметить, что для признания лекарственных препаратов взаимозаменяемыми в ряде случаев не требуется доказательств их биоэквивалентности. Например, это касается лекарственных препаратов, предназначенных для парентерального (подкожного, внутримышечного, внутривенного, внутриглазного, внутриполостного, внутрисуставного, внутрикоронарного) введения и представляющих собой водные растворы [16].
На сайте Государственного реестра лекарственных средств https://grls.rosminzdrav.ru/ размещается регулярно обновляемый перечень взаимозаменяемых лекарственных препаратов. Последний список представлен 04 июня 2021 г. [17].
Мексидол и его генерики с позиции современного законодательства
Рассмотрим, как реализуются данные требования на конкретном примере лекарственного препарата Мексидол (этилметилгидроксипиридина сукцинат). Мексидол — оригинальный лекарственный препарат, более 20 лет применяющийся в клинической практике [18, 19]. Эффективность и безопасность Мексидола были доказаны в ряде клинических исследований [20, 21], а сам препарат входит в Клинические рекомендации «Ишемический инсульт и транзи-торная ишемическая атака у взрослых» [22].
Учитывая данные обстоятельства, Мексидол широко применяется в клинической практике и количество его генериков постоянно увеличивается. По данным сайта «Государственный реестр лекарственных средств», на 25 августа 2021 г. в РФ было зарегистрировано 24 генерика Мексидо-ла (21 для лекарственной формы «раствор для инъекций» и 9 для пероральной лекарственной формы) [17].
Существенное различие в количестве воспроизведенных лекарственных препаратов для инъекционной и таблетированной лекарственных форм, скорее всего, связано с более простой процедурой регистрации инъекционной формы, для подтверждения биоэквивалентности которой не требуется выполнение клинических фармакокинетических исследований.
Препарат Мексидол по БКС относится к III классу [23], то есть хорошо растворимых, но подвергающихся ограниченной абсорбции лекарственным препаратом, поэтому регистрация его воспроизведенных лекарственных препаратов возможна по облегченной схеме, то есть только на основании исследований in vitro в тесте кинетики растворения («биовейвер»). Согласно сайту «Государственный реестр лекарственных средств», только для двух воспроизведенных препаратов этилметилгидроксипиридина сукцината были выполнены исследования in vivo и оценены сравнительная фармакокинетика и биоэквивалентность в клинических исследованиях [24, 25].
Также важно, что Мексидол — это лекарственный препарат, молекула которого состоит из двух частей: этилметилгидроксипиридина и сукцината. Этилметил-гидроксипиридин отвечает в молекуле за антиоксидантные свойства, обладая способностью связывать свободные радикалы [26, 27], а сукцинат обеспечивает антиги-поксический эффект этого препарата, являясь субстратом цикла Кребса, а также стимулирует специфические сук-цинатные рецепторы [28].
С этих позиций при проведении исследований по биоэквивалентности препарата Мексидол и его воспроизведенных лекарственных препаратов необходимо оценивать фармакокинетику не одного компонента — этилметилги-дроксипиридина, а двух — этилметилгидроксипиридина и сукцината, как рекомендовано для комбинированных лекарственных препаратов [29].
Заключение
С разработкой новых подходов и методов лечения различных заболеваний затраты на здравоохранение будут постоянно увеличиваться. Одним из подходов к снижению стоимости лечения является расширение использования генерических лекарственных препаратов. Однако принципиально важно, чтобы уменьшение стоимости лечения не сопровождалось снижением его качества. Согласно современным подходам, оптимальным является тестирование биоэквивалентности оригинального и воспроизведенного лекарственных препаратов в клинических исследованиях сравнительной фармакокинетики с последующей оценкой терапевтической эквивалентности.
Авторы заявляют об отсутствии конфликта интересов. The authors declare no conflicts of interest.
ЛИТЕРАТУРА/REFERENCES
1. Gupta R, Shah ND, Ross JS. Generic Drugs in the United States: Policies to Address Pricing and Competition. Clin Pharmacol Ther. 2019;105(2):329-337. https://doi.org/10.1002/cpt.1314
2. Kesselheim AS, Avorn J, Sarpatwari A. The high cost of prescription drugs in the United States: origins and prospects for reform. JAMA. 2016;316(8):858871. https://doi.org/10.1001/jama.2016.11237
3. Yu LX, Li BV. FDA Bioequivalence Standards. aapspress/Springer; 2014.
4. Lindenbaum J, Mellow MH, Blackstone MO, Butler VP Jr. Variation in biologic availability of digoxin from four preparations. N Engl J Med. 1971;285:1344-1347. https://doi.org/10.1056/NEJM197112092852403
5. Wagner JG, Christensen M, Sakmar E, et al. Equivalence lack in digoxin plasma levels. JAMA. 1973;224:199-204.
6. Skelly JP. Bioavailability and bioequivalence. J Clin Pharmacol. 1976;16:539-545. https://doi.org/10.1177/009127007601601013
7. Федеральный закон Российской Федерации №475 от 27 декабря 2019 г. «О внесении изменений в Федеральный закон «Об обращении лекарственных средств» и Федеральный закон «О внесении изменений в Федеральный закон «Об обращении лекарственных средств». Ссылка активна на 11.09.21. Federal’nyj zakon Rossijskoj Federacii №475 ot 27 dekabrja 2019 g. «O vnesenii izmenenij v Federal’nyj zakon «Ob obrashhenii lekarstvennyh sredstv» i Federal’nyj zakon «O vnesenii izmenenij v Federal’nyj zakon «Ob obrashhenii lekarstvennyh sredstv». (In Russ.). https://rg.ru/2019/12/31/a1804425-dok.html
8. Теоретические и практические основы проведения исследований воспроизведенных лекарственных препаратов: монография. Под ред. Хохлова А.Л. ООО Фотолайф; 2017. Teoreticheskie i prakticheskie osnovy provedenija issledovanij vosproizvedennyh lekarstvennyh preparatov: monografija. Pod red. Hohlova A.L. OOO Fotolajf; 2017. (In Russ.).
9. Решение №85 от 3 ноября 2016 г. «Об утверждении Правил проведения исследований биоэквивалентности лекарственных препаратов в рамках Евразийского экономического союза». Ссылка активна на 11.09.21. «Reshenie №85 ot 3 nojabrja 2016. «Ob utverzhdenii Pravil provedenija issledovanij biojekvivalentnosti lekarstvennyh preparatov v ramkah Evrazijskogo jekonomicheskogo sojuza». (In Russ.). https://docs.eaeunion.org/docs/ru-ru/01411942/cncd_21112016_85
10. Решение №67 от 4 сентября 2020 г. «О внесении изменений в Правила проведения исследований биоэквивалентности лекарственных препаратов в рамках в Евразийского экономического союза». Ссылка активна на 11.09.21. O vnesenii izmenenij v Pravila provedenija issledovanij biojekvivalentnosti lekarstvennyh preparatov v ramkah v Evrazijskogo ekonomicheskogo sojuza. (In Russ.). https://docs.eaeunion.org/docs/ru-ru/01427352/err_11092020_67
11. Draft Guidance. Bioequivalence Studies With Pharmacokinetic Endpoints for Drugs Submitted Under an ANDA Guidance for Industry. August 2021. U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER). https://www.fda.gov/media/87219/download
12. Hauschke D, Steinijans, Pigeot I. Bioequivalence studies in drug development. Methods and applications. Wiley; 2007.
13. Anderson S, Hauck WW Consideration of individual bioequivalence. J Pharmacokinet Biopharm. 1990;18:259-273. https://doi.org/10.1007/BF01062202
14. Hauck WW, Anderson S. Measuring switchability and prescribability: when is average bioequivalence sufficient? J Pharmacokinet Biopharm. 1994;22:551-556. https://doi.org/10.1007/BF02353794
15. Amidon GL, Lennernas H, Shah VP, Crison JR. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12:413-420. https://doi.org/10.1023/A:1016212804288
16. Федеральный закон Российской Федерации №429 от 22 декабря 2014 г. «О внесении изменений в Федеральный закон «Об обращении лекарственных средств»». Ссылка активна на 11.09.21. Federal’nyj zakon Rossijskoj Federacii №429 ot 22 dekabrja 2014 g. «O vnesenii izmenenij v Federal’nyj zakon «Ob obrashhenii lekarstvennyh sredstv». The link is active on 11.09.2021. (In Russ.). https://rg.ru/2014/12/26/lekarstva-dok.html
17. Государственный реестр лекарственных средств. Ссылка активна на 25 августа 2021. Gosudarstvennyj reestr lekarstvennyh sredstv. The link is active on August 25, 2021. https://grls.rosminzdrav.ru/Default.aspx
18. Воронина Т.А. Мексидол: спектр фармакологических эффектов. Журнал неврологии и психиатрии им. С.С. Корсакова. 2012;112(12):86-90. Voronina TA. Mexidol: the spectrum of pharmacological effects. Zhurnal Nevrologii i Psikhiatrii im. S.S. Korsakova. 2012;112(12):86-90. (In Russ.).
19. Щулькин А.В. Мексидол: современные аспекты фармакокинетики и фармакодинамики. Фарматека. 2016;4:65-71. Schulkin AV. Mexidol: modern aspects of the pharmacokinetics and pharmacodynamics. Farmateka. 2016;4:65-71. (In Russ.).
20. Скворцова В.И., Стаховская Л.В., Нарциссов Я.Р. и др. Рандомизированное двойное слепое плацебо-контролируемое исследование эффективности и безопасности мексидола в комплексной терапии ишемического инсульта в остром периоде. Журнал неврологии и психиатрии им. С.С. Корсакова (Приложение Инсульт). 2006;18:47-54. Skvortsova VI, Stakhovskaya LV, Nartsissov YaR, et al. Randomizirovannoe dvoinoe slepoe platsebo-kontroliruemoe issledovanie effektivnosti i bezopasnosti meksidola v kompleksnoi terapii ishemicheskogo insul’ta v ostrom periode. Zhurnal Nevrologii i Psikhiatrii im. S.S. Korsakova (Pril. Insul’t). 2006;18:47-54. (In Russ.).
21. Стаховская Л.В., Шамалов Н.А., Хасанова Д.Р. и др. Результаты рандомизированного двойного слепого мультицентрового плацебо-контролируемого в параллельных группах исследования эффективности и безопасности мексидола при длительной последовательной терапии у пациентов в остром и раннем восстановительном периодах полушарного ишемического инсульта (ЭПИКА). Журнал неврологии и психиатрии им. С.С. Корсакова. 2017;117:3(2):55-65. Stakhovskaya LV, Shamalov NA, Khasanova DR, et al. Results of a randomized double blind multicenter placebo-controlled, in parallel groups trial of the efficacy and safety of prolonged sequential therapy with mexidol in the acute and early recovery stages of hemispheric ischemic stroke (EPICA). Zhurnal Nevrologii i Psikhiatrii im. S.S. Korsakova. 2017;117:3(2):55-65. (In Russ.). https://doi.org/10.17116/jnevro20171173255-65
22. Клинические рекомендации. Ишемический инсульт и транзиторная ишемическая атака у взрослых. 2021. Ссылка активна на 11.09.21. Klinicheskie rekomendacii. Ishemicheskij insul’t i tranzitornaja ishemicheskaja ataka u vzroslyh. 2021. (In Russ.). https://cr.minzdrav.gov.ru/recomend/171_2
23. Гильдеева Г.Н. Формирование междисциплинарного подхода к стандартизации и контролю качества воспроизведенных лекарственных средств и преквалификационной экспертизе лекарственных препаратов: Дис. ... д-ра фарм. наук. М. 2017. Ссылка активна на 11 сентября 2021. Gil’deeva GN. Formirovanie mezhdisciplinarnogo podhoda k standartizacii i kontrolju kachestva vosproizvedennyh lekarstvennyh sredstv i prekvalifikacionnoj jekspertize lekarstvennyh preparatov: Dis. ... d-ra farm. nauk. M. 2017. (In Russ.). https://www.volgmed.ru/uploads/dsovet/thesis/2-766-gildeeva_geliya_ nyazyfovna.pdf
24. Сравнительное изучение биоэквивалентности лекарственных препаратов Нейрокс таблетки, покрытые пленочной оболочкой 125 мг, производства АО «ИИХР», Россия и Мексидол таблетки, покрытые пленочной оболочкой 125 мг, производства ЗАО «ЗиО-Здоровье», Россия. Ссылка активна на 11 сентября 2021. Sravnitel’noe izuchenie biojekvivalentnosti lekarstvennyh preparatov Nejroks tabletki, pokrytye plenochnoj obolochkoj 125 mg, proizvodstva AO «IIHR», Rossija i Meksidol tabletki, pokrytye plenochnoj obolochkoj 125 mg, proizvodstva ZAO «ZiO-Zdorov’e», Rossija. (In Russ.). https://grls.rosminzdrav.ru/ CIPermissionMi-ni.aspx? CIStatementGUID=29f8848a-4818-451f-b7d1-e0674c5d6372& CIPermGUID=13F402EE-6DC1-4A3D-9D76-7E1CB21B5557
25. Сравнительное изучение биоэквивалентности лекарственных препаратов Метостабил, таблетки, покрытые пленочной оболочкой, 125 мг, производства ООО «Озон», Россия и Мексидол, таблетки, покрытые оболочкой, 125 мг, производства «ЗиО-Здоровье», Россия. Ссылка активна на 11 сентября 2021. Sravnitel’noe izuchenie biojekvivalentnosti lekarstvennyh preparatov Metostabil, tabletki, pokrytye plenochnoj obolochkoj, 125 mg, proizvodstva OOO «Ozon», Rossija i Meksidol, tabletki, pokrytye obolochkoj, 125 mg, proizvodstva «ZiO-Zdorov’e», Rossija. The link is active on September 11, 2021. (In Russ.). https://grls.rosminzdrav.ru/CIPermissionMini.aspx? CIStatementGUID=d867af1a7fd14d91a9b4f0219325197f& CIPermGUID=C8E47357-82AA-4A48-8AEE-8A078D5CF62B
26. Щулькин А.В. Влияние мексидола на развитие феномена эксайтотоксичности нейронов in vitro. Журнал неврологии и психиатрии им. С.С. Корсакова. 2012;112(2):35-39. Shchul’kin AV. Effect of mexidol on the development of the phenomenon of the neuronal excitotoxicity in vitro. Zhurnal Nevrologii i Psikhiatrii im. S.S. Korsakova. 2012;112(2):35-39. (In Russ.).
27. Косолапов В.А., Спасов А.А., Анисимова В.А. Изучение антирадикальной активности новых соединений методами хемилюминесценции. Биомедицинская химия. 2005;51(3):287-294. Kosolapov VA, Spasov AA, Anisimova V. A Study of antiradical activity of new compounds by chemiluminescence. Biomeditsinskaya Khimiya. 2005;51(3):287-294. Accessed October 16, 2018. (In Russ.).
28. Кирова Ю.И., Германова Э.Л. Новые аспекты энерготропного действия мексидола. Патологическая физиология и экспериментальная терапия. 2018;62(4):42-46. Kirova YuI, Germanova EL. New aspects of the energy-tropic action of mexidol. Patologicheskaya Fiziologiya i Eksperimental’naya Terapiya. 2018;62(4):42-46. (In Russ.). https://doi.org/10.25557/0031-2991.2018.04.36-40
29. Рекомендация №25 от 2 сентября 2019 г. «О Руководстве по доклинической и клинической разработке комбинированных лекарственных препаратов». Ссылка активна на 11.09.21. Rekomendacija №25 ot 2 sentjabrja 2019 g. «O Rukovodstve po doklinicheskoj i klinicheskoj razrabotke kombinirovannyh lekarstvennyh preparatov». The link is active on 11.09.21. (In Russ.). https://docs.eaeunion.org/docs/ru-ru/01422928/clcr_05092019_25
Original and generic drugs: what does the clinician need to know?
© A.V. SHCHULKIN, A.A. FILIMONOVA
Ryazan State Medical University, Ryazan, Russia
Abstract
In the review article modern approaches to testing and registration of generic drugs are discussed. The article presents the history of the formation of the methodology for testing generic drugs and the current legislation of the Russian Federation. The stages of confirmation of equivalence of original and generic drugs are described: pharmaceutical equivalence, bioequivalence and therapeutic equivalence. The methods of assessing bioequivalence — as the main research in the registration of generic drugs — are discussed in detail. Using the example of the original neuroprotector — Mexidol (ethylmethylhydroxypyridine succinate) and its generics, it is described how legislative acts are implemented in practice. It is concluded that not all generic drugs are interchangeable for the original drug.
Keywords: original and generics drugs, ethylmethylhydroxypyridine succinate, mexidol.
Information about the authors:
Shchulkin A.V. — orcid.org/0000-0003-1688-0017
Filimonova A.A. — orcid.org/0000-0001-7524-3195
Corresponding author: Shchulkin A.V. — e-mail: alekseyshulkin@rambler.ru
To cite this article: Shchulkin AV, Filimonova AA. Original and generic drugs: what does the clinician need to know? S.S. Korsakov Journal of Neurology and Psychiatry = Zhurnal nevrologii ipsikhiatrii imeni S.S. Korsakova. 2021;121(10):99—104. (In Russ.). doi.org/10.17116/jnevro202112110199
Комментарии
ПРАКТИКА ПЕДИАТРА



