Особенности развития неврологических осложнений у пациентов с сахарным диабетом 2-го типа и метаболическим синдромом: возможность коррекции и профилактики
СтатьиОпубликовано в журнале:
«Терапевтический архив», 1, 2015.
В.Н. Шишикова
ГБУЗ «Центр патологии речи нейрореабилитации» ДЗ Москвы, Москва, Россия
Аннотация
Распространенность сахарного диабета (СД) 2-го типа и метаболических нарушений, ему предшествующих, достигла масштабов эпидемии. Окислительный стресс играет значительную роль в развитии микро- и макрососудистых осложнений у больных СД. Накопление свободных радикалов отвечает за активацию патологических биохимических путей, ответственных за развитие системного и сосудистого воспаления, дисфункции эндотелия, гиперкоагуляционных и ишемических состояний. Поскольку повреждение сосудистой и нервной систем не нивелируется даже при адекватном контроле гликемии, необходимы комплексные патогенетические стратегии лечения. Одним из обязательных компонентов комплексной терапии осложнений СД является применение антиоксидантной терапии с помощью препарата мексидол.
Ключевые слова: сахарный диабет 2-го типа, метаболический синдром, окислительный стресс, мексидол.
Specific features of neurological complications developing in patients with type 2 diabetes mellii and metabolic syndrome: Possibility for correction and prevention
V.N. Shishkova
Center for Speech Pathology and Neurorehabilitation, Moscow Healthcare Department, Moscow, Russia
The prevalence of type 2 diabetes mellitus (DM) and preceding metabolic disturbances has reached epidemic proportions. Oxidative stress plays a significant role in the development of micro- and macrovascular complications in patients with DM. The accumulation of free radicals is responsible for the development of systemic and vascular inflammation, endothelial dysfunction, and hypercoagulable and ischemic states. Since vascular and nervous system damages do not level off even under adequate glycemic control, there is a need for complex pathogenetic treatment strategies. Antioxidant therapy using mexidol is one of the compulsory components of combination therapy for complications of DM.
Key words: type 2 diabetes mellitus, metabolic syndrome, oxidative stress, mexidol.
ATII — ангиотензин II
АГ — артериальная гипертония
АД — артериальное давление
ГИ — гиперинсулинемия
ГМК — гладкие мышечные клетки
ДН — диабетическая невропатия
ДПН — диабетическая полиневропатия
ДЭ — дисфункция эндотелия
ДЭП — диабетическая энцефалопатия
ИР — инсулинорезистентность
КН — когнитивные нарушения
МАРК — митогенактивированная протеинкиназа
МС — метаболический синдром
ПС-ВНС — парасимпатическая часть вегетативной нервной системы
РАС — ренин-ангиотензиновая система
С-ВНС — симпатическая часть вегетативной нервной системы
СДСП — симметричная дистальная сенсорная полинейропатия
СЖК — свободные жирные кислоты
ССЗ — сердечно-сосудистые заболевания
ХИМ — хроническая ишемия мозга
ЦНС — центральная нервная система
В последние десятилетия ученые стали рассматривать различные метаболические нарушения или заболевания, ассоциированные с избыточной массой тела и центральным типом ожирения, в комплексе и высказывать предположения об общности этих процессов. В 1988 г. американский ученый G. Reaven, объединив нарушения углеводного обмена, артериальную гипертонию (АГ) и дислипидемию понятием «синдром Х», впервые высказал предположение о том, что объединяющей основой этих нарушений могут быть инсулинорезистентность (ИР) и компенсаторная гиперинсулинемия (ГИ). В конце прошлого века метаболические нарушения и заболевания, развивающиеся у лиц с ожирением, объединили в понятие «метаболический синдром» (МС). Таким образом, МС — это сочетание метаболических нарушений, в патогенезе которых важную роль играет ИР, потенцирующая раннее развитие атеросклероза и его осложнений [1].
Существует 5 групп диагностических критериев МС. Отечественными учеными, экспертами Всероссийского научного общества кардиологов также разработаны и опубликованы критерии МС. Наличие у пациента центрального ожирения и двух дополнительных критериев служит основанием для диагностирования МС.
К критериям МС относятся следующие:
— абдоминальное ожирение — объем талии ≥94 см у мужчин и >80 см у женщин;
— уровень глюкозы в крови натощак >5,6 ммоль/л;
— артериальное давление (АД) ≥130/85 мм рт.ст.;
— уровень липопротеидов высокой плотности <1,03 ммоль/л у мужчин и <1,29 ммоль/л у женщин;
— уровень триглицеридов в крови >1,69 ммоль/л.
ИР — взаимосвязь атеросклероза, АГ и СД 2-го типа. Согласно современным представлениям ключевым звеном патогенеза МС является первичная ИР и компенсаторная ГИ.
ИР — нарушение опосредованной инсулином утилизации глюкозы клетками, которое сопровождает целый ряд физиологических и патологических процессов. Физиологическая ИР выявляется в пубертатном периоде, при беременности, в климактерическом периоде, во время ночного сна, после приема богатой жиром пищи [1].
Метаболическая ИР характерна для МС, СД 2-го типа, декомпенсированного СД 1 -го типа, диабетического кетоацидоза, ожирения, выраженной недостаточности питания, гиперурикемии, гипогликемии, индуцированной инсулином, злоупотребления алкоголем. Эндокринная ИР отмечается при таких заболеваниях, как гипотиреоз, синдром Кушинга, акромегалия, феохромоцитома. Неэндокринная ИР типична для гипертонической болезни, хронической почечной недостаточности, цирроза печени, сердечной недостаточности, ревматоидного артрита, черного акантоза, миотонической дистрофии, травм, ожогов, сепсиса, состояния после хирургических вмешательств, раковой кахексии [2].
Наибольшее клиническое значение при ИР имеет потеря чувствительности к инсулину мышечной, жировой и печеночной тканями. Предполагают, что причиной ускоренного атерогенеза и высокой летальности от ишемической болезни сердца и инсультов у больных СД 2-го типа также могут быть ИР и сопутствующая ей ГИ. У пациентов с ИР имеются дефекты генов, ответственных за передачу сигнала после соединения инсулина со своим рецептором (пострецепторные дефекты), и прежде всего у них нарушаются транслокация и синтез внутриклеточного транспортера глюкозы ГЛЮТ-4. Кроме того, возможны генетические дефекты на уровне субстрата рецептора инсулина 1-го типа и/или фосфатидилинозитол-3-киназы; обнаружены также нарушения экспрессии других генов, обеспечивающих метаболизм глюкозы и липидов: глюкозо-6-фосфатдегидрогеназы, глюкокиназы, липопротеинлипазы, синтазы жирных кислот и др. Генетическая предрасположенность к ИР может не реализоваться и не проявиться клинически (в виде МС и/или СД 2-го типа) в отсутствие необходимых для этого внешних факторов: избыточного калорийного питания (особенно жирной пищи) и низкой физической активности. Эти факторы сами способствуют увеличению абдоминального ожирения, накоплению свободных жирных кислот (СЖК) и, следовательно, усилению имеющейся ИР [1, 2].
Развивающаяся при ИР компенсаторная ГИ, с одной стороны, позволяет в начале поддерживать углеводный обмен в норме, с другой — способствует развитию метаболических, гемодинамических и органных нарушений, приводящих в конечном итоге к развитию сердечно-сосудистых заболеваний (ССЗ) и СД 2-го типа [3].
ИР мышечной ткани проявляется в снижении поступления глюкозы из крови в миоциты и ее утилизации в мышечных тканях. ИР жировой ткани проявляется в резистентности к антилиполитическому действию инсулина, приводящему к накоплению СЖК и глицерина. СЖК поступают в печень, где становятся основным источником формирования атерогенных липопротеинов очень низкой плотности. ИР ткани печени характеризуется снижением синтеза гликогена и активацией процессов распада гликогена до глюкозы (гликогенолиз) и синтеза глюкозы de novo из аминокислот, лактата, пирувата, глицерина (глюконеогенез), в результате чего кислота из печени поступает в кровоток [3].
В целом ИР — это эволюционно закрепленный механизм выживания в неблагоприятных условиях, когда периоды изобилия чередовались с периодами голода. Наличие ИР обеспечивало накопление энергии в виде отложений жира, запасов которого хватало на то, чтобы пережить голод. В современных условиях в странах с высоким экономическим развитием, постоянно сопутствующим изобилием и склонностью к малоподвижному образу жизни сохранившиеся в генетической памяти механизмы ИР продолжают «работать» на накопление энергии, что способствует развитию абдоминального ожирения, дислипидемии, раннему атеросклерозу, АГ и СД 2-го типа [1—3].
Существенную роль играет ИР в развитии АГ. Взаимосвязь ГИ (маркера ИР) и эссенциальной АГ настолько прочна, что при высокой концентрации инсулина в плазме у больного можно прогнозировать развитие в скором времени АГ. Причем эта связь прослеживается как у больных с ожирением, так и у лиц с нормальной массой тела. Существует несколько механизмов, объясняющих повышение АД при ГИ [4].
Инсулин способствует активации симпатической части вегетативной нервной системы (С-ВНС), повышению АД при ГИ. Инсулин способствует активации С-ВНС, повышению реабсорбции натрия и воды в почечных канальцах, внутриклеточному накоплению натрия и кальция, инсулин как митогенный фактор активирует пролиферацию гладких мышечных клеток (ГМК) сосудов, что ведет к утолщению их стенки. Механизм влияния инсулина на С-ВНС до конца неясен. Предполагают, что инсулин может активировать С-ВНС путем прямого воздействия на центральную нервную систему (ЦНС), проникая через гематоэнцефалический барьер в перивентрикулярную область гипоталамуса, где, связываясь со своими рецепторами на поверхности нейронов, блокирует активность парасимпатической части вегетативной нервной системы (ПС-ВНС) и, напротив активирует С-ВНС. G. Reaven основоположник учения о синдроме ИР, предположил, что причиной гиперактивации С-ВНС в условиях гипергликемии может быть повышен метаболизм глюкозы в ядрах гипоталамуса, а это тормозит передачу блокирующих импульсов на симпатические центры продолговатого мозга [4].
Стимуляция С-ВНС при ГИ сопровождается увеличением сердечного выброса, повышением общего периферического сосудистого сопротивления, что неизбежно приводит к повышению АД. Одновременное снижение активности ПС-ВНС, вызванной ГИ, увеличивает частоту сердечных сокращений. Повышение реабсорбции натрия и воды происходит также под влиянием ГИ. Инсулин оказывает прямое воздействие на проксимальные канальцы нефронов, повышая реабсорбцию натрия и жидкости. Помимо антинатрийуреза инсулин вызывает антикалийурез и антиурикозурию. В результате увеличивается объем циркулирующей жидкости, что приводит к повышению сердечного выброса [3, 4].
Внутриклеточное накопление натрия и кальция — эффект действия инсулина. Инсулин блокирует активность Na/K- и Ca/ Mg-АТФазы клеточных мембран, что приводит к повышению внутриклеточного содержания натрия и кальция. Вследствие накопления этих электролитов в стенке сосудов повышается чувствительность сосудистых рецепторов к действию сосудосуживающих факторов. Под влиянием инсулина происходит утолщение стенки сосудов. Митогенные свойства инсулина обнаружены достаточно давно в серии экспериментальных работ, в которых показано, что инсулин стимулирует рост, пролиферацию и миграцию ГМК сосудов, приводит к утолщению их стенки. В норме инсулин, связываясь с рецепторами на поверхности клеток эндотелия, может действовать двумя различными путями. Первый путь — активация секреции оксида азота (NO) через субстраты инсулиновых рецепторов и фосфатидилинозитол-3-киназу. Этот механизм обеспечивает сосудорасширяющие и антиатерогенные свойства инсулина, участвует в инсулинзависимом транспорте глюкозы в клетки. Второй путь — реализация митогенных свойств инсулина через каскад посредников, повышающих активность митогенактивированной протеинкиназы (МАРК), что завершается пролиферацией и миграцией ГМК, активацией синтеза сосудосуживающего фактора эндотелина-1 и повышением АД. Оказалось, что в условиях ИР первый механизм не работает — именно этот путь резистентен к действию инсулина, следовательно, молекула NO не синтезируется. В то же время второй механизм сохраняет свою высокую активность. Поэтому ГИ, развивающаяся вследствие ИР (при МС, СД 2-го типа, висцеральном ожирении), не только не снижает АД, а напротив, оказывает прогипертензивное и атерогенное действие [3, 4].
Существует взаимосвязь активности ренин-ангиотензиновой системы (РАС), уровня АД, и чувствительности тканей к инсулину. Хорошо известно, что гиперактивность РАС стойко поддерживает высокое АД. Однако лишь недавно, в экспериментальных условиях, получены убедительные данные о том, что ангиотензин II (ATII) дозозависимо ингибирует пострецепторную сигнальную систему инсулина, реализующую транспорт глюкозы в клетки и продукцию NO. Одновременно ATII стимулирует МАРК, задействованную в осуществлении митогенной и пролиферативной активности инсулина [5]. Следовательно, гиперактивность РАС и ATII в свою очередь вызывает резистентность тканей к антиатерогенному и гипотензивному действию инсулина, что приводит к развитию АГ, прогрессии ССЗ а также блокирует транспорт глюкозы в клетки, что способствует развитию СД 2-го типа.
Таким образом, описанные механизмы, объединяющие патогенез развития СД 2-го типа и ССЗ, дают представление о масштабе микро- и макрососудистых осложнений СД 2-го типа, формирующихся одновременно с началом заболевания.
Хроническая ишемия мозга (ХИМ) у больных СД 2-го типа.
Наряду с другими осложнениями СД структурные и функциональные изменения нервной системы играют роль фактора, снижающего качество и ограничивающего продолжительность жизни больных СД. СД 2-го типа — наиболее важный фактор риска развития ишемических инсультов и транзиторных ишемических нарушений в головном мозге. Относительный риск развития инсульта выше у лиц с СД 2-го типа в 1,8—6 раз по сравнению с таковым у лиц без СД. В исследовании MRFIT риск смерти от инсульта среди пациентов с СД был в 2,8 раза выше, чем у пациентов без СД, при этом риск смерти от ишемического инсульта был выше в 3,8 раза, от субарахноидального кровоизлияния — в 1,1 раза и внутримозгового кровоизлияния — в 1,5 раза [6].
СД является фактором риска развития нарушений мозгового кровообращения независимо от наличия других факторов риска (АГ и повышение уровня холестерина) [6]. В развитии инсульта при СД ведущая роль принадлежит ХИМ. Существенную роль в развитии ХИМ при СД играет патология магистральных артерий: сонных и позвоночных, которые при СД быстро поражаются атеросклерозом. Показано, что СД и гипергликемия без СД (например, преддиабет) являются независимыми факторами риска развития системного атеросклероза с поражением сосудов различных локализаций, в том числе мозговых [7]. Кроме того, для СД характерно системное поражение сосудов микроциркуляторного русла (микроангиопатия), которое сопровождается развитием нарушений микроциркуляции в органе-мишени, т.е. в том числе в головном мозге. Микроангиопатия мозга усугубляет метаболические нарушения, развивающиеся при ХИМ, и повышает риск развития деменции, при этом отмечается значимое повышение риска развития болезни Альцгеймера [8].
Дополнительная проблема при СД — компенсация уровня глюкозы в крови, которая связана с риском возникновения состояний резкой гипогликемии (уровень глюкозы в крови ниже нормы). Известно, что гипогликемический индекс нарастает с длительностью СД более 6 лет, при этом у пациентов состояния выраженной гипогликемии ассоциированы с высоким риском деменции, а дополнительный риск деменции при сравнении лиц без гипогликемических эпизодов и пациентов с зарегистрированными эпизодами составил 2,39% в год [8]. Многочисленные публикации свидетельствуют, что при СД гораздо раньше, чем при изолированной АГ или атеросклерозе, отмечаются снижение скорости психомоторных реакций, нарушение функции лобной доли, снижение памяти, комплексные моторные нарушения, снижение внимания и другие клинические проявления ХИМ [8—11].
Таким образом, тяжесть повреждений ЦНС при СД определяется степенью и длительностью снижения мозгового кровотока (обусловленное атеросклерозом и/или АГ), а также нарушениями метаболизма в головном мозге.
СД 2-го типа — причина когнитивных нарушений (КН).
Одним из клинически значимых независимых факторов риска развития когнитивных нарушений (КН) и деменции в настоящее время является СД 2-го типа. При этом сопутствующая СД сердечно-сосудистая патология определяет лишь более выраженное снижение психической активности и интеллектуальной гибкости, что отражает более выраженную дисфункцию структур головного мозга [9]. КН не всегда бывают следствием только структурного поражения головного мозга, их развитие может быть обусловлено метаболическими расстройствами или возможно сочетания нескольких патологических факторов (сосудистый и метаболический) [10]. Увеличение распространенности и тяжести КН с течением времени у пациентов с СД 2-го типа связано в первую очередь с прогрессированием ХИМ [11].
Исследования показали, что в дебюте СД 2-го типа почти у 50% больных имеются макро- и микрососудистые осложнения. Возможно, это результат того, что метаболические нарушения возникают гораздо раньше первых клинических проявлений СД, и к моменту постановки диагноза приводят к необратимым сосудистым изменениям [9, 10]. Наиболее драматично развитие центральных форм диабетической невропатии (ДН), к которым относятся острые нервно-психические расстройства на фоне декомпенсации метаболизма (кетоацидоза, лактатацидоза гиперосмолярного и гипогликемического состояний), острые нарушения мозгового кровообращения — инсульт, преходящие нарушения мозгового кровообращения; прогрессирующая диабетическая энцефалопатия (ДЭП). К истинно диабетической принято относить прогредиентно развивающуюся на фоне нарушений углеводного обмена метаболическую энцефалопатию [10]. Однако выделение исключительно метаболической формы энцефалопатии при СД весьма проблематично, поскольку с течением заболевания прогрессируют нарушения мозга, обусловленные развитием сосудистых изменений, прогрессией атеросклероза, АГ и автономной невропатии.
ДЭП обычно развивается исподволь, у молодых пациентов ее хронические проявления усугубляются последствиями перенесенных острых гипер- и гипогликемических эпизодов, у пожилых — нарушениями мозгового кровообращения [11].
Как и при других метаболических энцефалопатиях, клинические проявления ДЭП неспецифичны, наиболее часто развивается нарушение когнитивных функций: снижение памяти и внимания, замедление мышления, снижение скорости психических реакций и способности к обучению. Эти проявления ДЭП очень важны для правильного понимания всех трудностей взаимодействия с такими пациентами в лечебном процессе [10, 11].
Установлена общность механизмов развития когнитивного дефицита при СД и болезни Альцгеймера, больные с СД входят в группу риска развития деменции. Известно, что в процессе старения мозга принимают участие те же патогенетические звенья, что и при развитии осложнений СД, ключевым из которых является окислительный стресс [12].
Окислительный стресс при СД может быть следствием следующих механизмов:
— повышенного образования свободных радикалов, образующихся при окислении как самих углеводов, так и углеводов, образующих комплексы с различными белками, а также в результате аутоокисления жирных кислот в триглицеридах, фосфолипидах и эфирах холестерина;
— снижения активности антиоксидантной системы в организме, которая представлена глутатионом, глутатионпероксидазой, каталазой, супероксиддисмутазой, витаминами К, Е, С, а-липоевой кислотой и другими антиоксидантами (таурин, каротин, мочевая кислота и коэнзим Q10);
— нарушения ферментов полиолового обмена глюкозы, митохондриального окисления, обмена простагландинов и лейкотриенов и снижения активности глиоксалазы;
— нарушения концентрации или обмена глутатиона и ионов некоторых металлов.
Кроме того, ишемия, гипоксия и псевдогипоксия тканей, наблюдаемая при СД, являются дополнительными факторами, способствующими повышенному образованию свободных радикалов в различных органах и тканях [13].
Выявляемые при нейропсихологическом обследовании изменения у больных СД 2-го типа более стойкие, чем при СД 1-го типа. Это чаще всего средней степени выраженности нарушения вербальной памяти и процесса обработки информации, также при СД выявляются нарушения праксиса, гнозиса, речевых и пространственных функций, зрительной и слуховой памяти, а также нарушения межполушарных взаимодействий с дисфункцией правого полушария [13, 14].
Течение СД сопровождается частыми колебаниями уровня глюкозы в крови, порой резкими, которым придается очень большое значение в развитии расстройств ЦНС. Особенно опасны в этом отношении эпизоды гипогликемии, возникающие во время терапии инсулином или пероральными сахароснижающими средствами, стимулирующими выработку инсулина.
Когнитивный дефицит часто наблюдается и у больных с впервые диагностированным СД 2-го типа, еще не получавших сахароснижающую терапию, но у которых диагностируется энцефалопатия. Это состояние, по мнению многих ученых, при СД является неизбежным [14]. Патогенез ДЭП представляет собой многофакторный процесс, сходный с таковым при диабетической полиневропатии (ДПН), когда в патогенез вовлекается дисфункция сосудистого эндотелия, приводящая к уменьшению кровоснабжения нервов и мозговой ткани, нарушение трофики и прямое токсическое влияние гипергликемии на нервы. Влияние СД на мозг более выражено у людей среднего и пожилого возраста и приводит к ускорению обусловленного старением когнитивного снижения и развитию энцефалопатии [12—14].
Особым вопросом в общей проблеме СД является влияние инсулина на когнитивные функции. Инсулин, идентичный панкреатическому, и типичные инсулиновые рецепторы широко экспрессируются в разных отделах мозга, в частности в височных долях. Инсулин проникает в мозг посредством рецепторзависимого транспорта через гематоэнцефалический барьер и принимает участие в регуляции энергетического гомеостаза, репродуктивных и когнитивных функций организма. Вопросы, касающиеся нейрональных эффектов инсулина, остаются во многом дискуссионными, однако в последнее десятилетие получены убедительные доказательства того, что инсулин и инсулинорецепторная сигнальная система мозга необходимы для нормального функционирования нейронов. Дисфункция этой системы приводит к развитию нейродегенеративных заболеваний [15]. Показано, что инсулин и инсулиновые рецепторы играют важную роль в синаптической передаче и могут быть связаны с такими важнейшими функциями мозга, как пищевое поведение, обучение и память. Имеются данные о том, что инсулин может принимать непосредственное участие в реализации ряда когнитивных функций, а нарушения его метаболизма могут сопровождаться возникновением ряда неврологических синдромов и когнитивных расстройств. Гиперинсулинемия может определять когнитивное снижение, а нарушения в системе синтеза и секреции инсулина негативно влиять на когнитивные функции [16].
Помимо выполнения в головном мозге медиаторных функций инсулин принимает участие в регуляции синтеза белка — предшественника амилоида и продукта его метаболизма β-амилоида — основного компонента амилоидных отложений, а также регулирует фосфорилирование τ-протеина, составляющего основу нейрофибриллярных образований [15, 16]. Таким образом, можно говорить о патогенетической связи СД 2-го типа не только с сосудистым поражением головного мозга, но и с нейродегенеративным процессом, лежащим в основе деменции. В настоящее время нет клинических исследований, в которых доказана эффективность любых сахароснижающих препаратов, применяемых в современной терапии СД, особенно 2-го типа, в предотвращении именно КН, что делает вопрос профилактики развития этих состояний очень актуальным [17].
ДПН.
Это патогенетически связанное с СД сочетание синдромов поражения нервной системы, классифицируемое в зависимости от преимущественного поражения спинномозговых нервов (дистальная или периферическая невропатия) и вегетативной нервной системы (висцеральная или автономная невропатия).
ДН — самое частое осложнение СД. В среднем частота выявления невропатии среди пациентов с СД составляет 25%, при углубленном неврологическом исследовании она возрастает до 50%, а при применении электрофизиологических методов исследования, исследовании вегетативных функций и количественной оценки чувствительности — до 90%. ДПН — обычно позднее осложнение у пациентов с СД 1-го типа, однако у пациентов с СД 2-го типа невропатия может быть первой диагностической находкой [18]. В связи с длительным латентным периодом СД 2-го типа частой клинической ситуацией может быть выявление признаков ДПН, предшествующих появлению признаков самого СД. У многих пациентов неврологические нарушения являются первыми симптомами заболевания, даже на этапе начальных нарушений углеводного обмена — нарушенной толерантности к глюкозе и гипергликемии натощак и позволяют в последующем, при своевременном проведении лечебно-профилактических мероприятий, предотвратить развитие СД. С увеличением длительности и тяжести СД частота ДПН неуклонно возрастает [19].
Классификация ДН затруднена, так как часто имеется сочетание нескольких синдромов. Ряд авторов классифицируют ДН в зависимости от преимущественного вовлечения в процесс спинномозговых нервов (периферическая невропатия) и/или вегетативной нервной системы (автономная невропатия). Одним из наиболее распространенных клинических вариантов поражения периферической нервной системы является симметричная дистальная сенсорная полинейропатия (СДСП) с преимущественным поражением нервов нижних конечностей. СДСП имеет неблагоприятное прогностическое значение в отношении формирования диабетической стопы, высокого риска ампутации конечности, наступления летального исхода. Этот тип невропатии, как правило, отражает длительность и тяжесть гипергликемии и зависит от продолжительности СД и эффективности гипогликемической терапии, но иногда бывает первым проявлением скрыто протекавшего СД. Кроме того, этот вариант ДН часто сочетается с другими микрососудистыми осложнениями СД — нефропатией и ретинопатией. Гипергликемия, обусловленная СД, вызывает такие серьезные метаболические нарушения, как избыточное гликирование белков и окислительный стресс, которые существенно нарушают структуру и функции нейронов [20]. Окислительный стресс способствует повреждению эндотелиальных клеток, лежащих в основе мембраны эндоневральных сосудов, что приводит к микроваскулярной дисфункции. Развивающиеся в итоге гипоксия и ишемия еще в большей степени активируют процессы поражения нервов. Существенным механизмом формирования ДПН является дефицит нейротрофических факторов, снижающий способности пораженного нерва к регенерации. В патогенезе определенную роль играют также активация протеинкиназы С и блокирование выделения оксида азота, регулирующего тонус сосудов, что способствует развитию дисфункции эндотелия (ДЭ) и прогрессированию микроангиопатии. Появление боли при ДПН обусловлено поражением тонких сенсорных волокон, ответственных за болевую чувствительность [21].
Возможность коррекции и профилактики неврологических осложнений у пациентов с СД и МС.
Окислительный стресс играет значимую роль в развитии микро- и макрососудистых осложнений у больных СД. Накопление свободных радикалов в сосудах при СД отвечает за активацию патологических биохимических путей, ответственных за развитие системного и сосудистого воспаления, ДЭ, гиперкоагуляционных и ишемических состояний (см. рисунок).
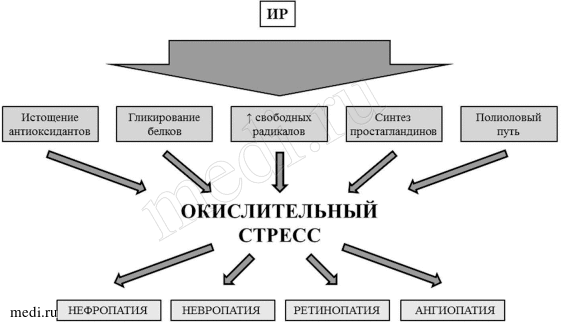 Осложнения СД.
Осложнения СД.
Поскольку повреждение сосудистой и нервной системы не нивелируется даже при адекватном контроле гликемии, необходимы комплексные патогенетические стратегии лечения. Одним из обязательных компонентов комплексной терапии диабетических осложнений является применение антиоксидантной терапии, которую по праву можно отнести к патогенетической, так как роль участия свободных радикалов в патогенезе СД и его осложнений в настоящее время не вызывает сомнений [2, 13].
Современная антиоксидантная терапия представлена различными препаратами, которые широко применяются не только в комплексном лечении больных СД, но и других системных заболеваний. Известны прямые антиоксиданты, которые сами связывают свободные радикалы, и непрямые антиоксиданты, стимулирующие собственную антиоксидантную защиту. Кроме того, антиоксиданты делят на природные (эндогенные) и синтетические. Механизм и степень выраженности антиоксидантного действия различных соединений зависит от того, в какой среде или структуре он реализует свой антиоксидантный эффект. Так, эндогенные антиоксиданты, к которым, например, относится витамин С, обладают наибольшей растворимостью в полярных растворителях и реализуют свое антиоксидантное действие только в плазме, межклеточной жидкости и на внеклеточном уровне [13]. Плазматический слой клеточной мембраны, состоящий из фосфолипидов, может быть защищен от реактивных соединений веществами иной структуры, из которых наиболее эффективным является этилметилгидроксипиридина сукцинат, хорошо известный под торговым названием «мексидол».
Механизм действия мексидола обусловлен его главными свойствами: антиоксидантным и мембранопротекторным. Он ингибирует процессы перекисного окисления липидов, повышает собственную защитную активность супероксидоксидазы, уменьшает вязкость мембраны и стабилизирует ее [22]. В ЦНС мексидол модулирует активность связанных с мембраной ферментов (кальцийнезависимой фосфодиэстеразы, аденилатциклазы, ацетилхолинэстеразы), рецепторных комплексов (бензодиазепинового, ГАМК, ацетилхолинового). Это усиливает их способность связывания с лигандами, способствует сохранению структурно-функциональной организации биомембран, транспорта нейромедиаторов и улучшению синаптической передачи, что является фундаментальным принципом улучшения функционирования нейрональной передачи и сохранения когнитивных функций. Под действием этого препарата повышается устойчивость организма к стрессу, при этом отмечается и анксиолитическое действие препарата, не сопровождающееся сонливостью и миорелаксирующим эффектом. Мексидол также обладает ноотропными свойствами, предупреждает и уменьшает нарушения обучения и памяти, возникающие при старении и патологических состояниях, обладает антигипоксическими свойствами, повышает концентрацию внимания и работоспособность в условиях ХИМ [23, 24]. Препарат также улучшает метаболизм в ЦНС, микроциркуляцию и реологические свойства крови, уменьшает агрегацию тромбоцитов, стабилизирует мембранные структуры клеток крови (эритроцитов и тромбоцитов) [22, 23]. Накоплен клинический опыт применения мексидола у пациентов с МС и СД [25—29]. Так, в проведенном О.В. Занозиной и соавт. [25] крупном исследовании подтверждены метаболические, гемореологические, гемодинамические нарушения у больных СД 2-го типа даже при его компенсации. Доказана ведущая роль активации процессов перекисного окисления липидов и снижения активности антиоксидантной системы в развитии поздних осложнений при СД. Авторами определена эффективная доза антиоксидантного препарата мексидол, доказана его эффективность на молекулярном и клеточном уровнях, позволяющая врачу дифференцированно назначать данный препарат при лечении больных СД.
Применение мексидола в 4-недельной комплексной терапии у пациентов с МС привело к уменьшению выраженности ДЭ, снижению гиперхолестеринемии и гипертриглицеридемии, а также уровня протромбина и фибриногена, что способствовало улучшению микроциркуляции. Выявлена способность мексидола снижать ГИ у пациентов с ИР, особенно важно снижение активности аланинаминотрансферазы, косвенно отражающее состояние печеночной ИР. Отмечены безопасность мексидола и его эффективное сочетание с другими лекарственными средствами в комплексной терапии МС [26]. Сходные данные получены авторами в недавнем исследовании по оценке эффективности антиоксидантов у пациентов с МС, клинически значимыми оказались результаты на фоне терапии мексидолом: снижение гликемии, гипертриглицеридемии и ИР [27].
Высокая оценка эффективности терапии мексидолом КН разной степени выраженности у пациентов с СД дана в длительном наблюдательном исследовании [28]. Автором отмечено достоверное улучшение когнитивных функций после терапии мексидолом независимо от длительности течения СД и применяемых гипогликемических препаратов.
В профилактике и терапии осложнений СД отдельно выделяется направление, связанное с ДН, а именно с СДСП, так как именно она является началом такого грозного осложнения, как синдром диабетической стопы и часто ведет к ампутации конечности вследствие гангрены. В исследованиях проф. И.А. Волчегорского и соавт. [29, 30] применение мексидола внутривенно капельно у пациентов с СДСП и синдромом диабетической стопы привело к клинически значимому улучшению объективных показателей клинического неврологического статуса и сопровождалось редукцией симптоматики ДН. Мексидол вызывал значительное, более чем двукратное, снижение суммарной оценки по шкале невропатического симптоматического счета, у пациентов уменьшилась выраженность таких мучительных симптомов ДН, как жжение, онемение и судороги в дистальных отделах нижних конечностей. Схема назначения мексидола пациентам с неврологическими осложнениями СД следующая: 500 мг в сутки внутривенно капельно 10—14 дней с последующим переходом на таблетированную форму по 1—2 таблетки (125—250 мг) 3 раза в день в течение 1—3 месяцев.
В заключение следует подчеркнуть, что лечение каждого пациента с СД должно быть индивидуальным с учетом клинических особенностей и возможных имеющихся осложнений, а также наличия сочетанных сосудистых заболеваний. При выборе антиоксидантных лекарственных препаратов, помимо непосредственного антиоксидантного эффекта, должны быть учтены другие положительные эффекты каждого препарата, а также его переносимость и возможность безопасного сочетания с другими лекарствами в составе комплексной терапии. Этими качествами обладает современный эффективный антиоксидант с мембранопротективным, ноотропным, вегетостабилизирующим и метаболическим свойствами мексидол.
Литература
1. DeFronzo R.A., Ferrannini E. Insulin resistance. A multifaceted syndrome responsible for NIDDM, obesity, hypertension, dyslipidemia, and atherosclerotic cardiovascular disease. Diabetes Care 1991; 14 (3): 173-194.
2. Дедов И.И., Шестакова М.В. Сахарный диабет. М: Медицинское информационное агентство 2005; 677.
3. Rosenzweig J.L., Ferrannini E., Grundy S.M. et al. Primary prevention of cardiovascular disease and type 2 diabetes in patients at metabolic risk: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2008 93 (10): 3671-3689.
4. Brands J.W. Obesity and Hypertension: Roles of Hyperinsulinemia, Sympathetic Nervous System and Intrarenal Mechanisms. J Nutr 1995; 125 (6 Suppl): 1725S-1731S.
5. Nigro J., Osman N, Dart A. et al. Insulin resistance and atherosclerosis. Endocrine Rev 2006; 27 (3): 242-259.
6. Stamler J., Vaccaro O., Neaton J.D. et al. Diabetes, other risk factors, and 12-yr cardiovascular mortality for men screened in the Multiple Risk Factor Intervention Trial. Diabetes Care 1993; 16: 434-444.
7. Stegmayr B., Asplund K. Diabetes as a risk factor for stroke. A population perspective. Diabetologia 1995; 38: 1061-1068.
8. Stewart R., Liolitsa D. Type 2 diabetes mellitus, cognitive impairment and dementia. Diabetic Med 1999; 16 (2): 93-112.
9. Захаров В.В., Сосина В.Б. Когнитивные нарушения у больных сахарным диабетом. Неврол журн 2009; 4: 54-58.
10. Калинин А.П., Котов С.В. Неврологические расстройства при эндокринных заболеваниях. М: Медицина 2001; 272.
11. Vanhanen M., Koivisto K., Karjalainen L. et al. Risk for non-insulin-dependent diabetes in the normoglycaemic elderly is associated with impaired cognitive function. Neuroreport 1997; 8 (6): 1527-1530.
12. Kalmijn S., Feskens E.J., Launer L.J. et al. Glucose intolerance, hyperinsulinaemia and cognitive function in a general population of elderly men. Diabetologia 1995; 38 (9): 1096-1102.
13. Балаболкин М.И., Креминская В.М., Клебанова Е.М. Роль окислительного стресса в патогенезе диабетической нейропатии и возможность его коррекции препаратами α-липоевой кислоты. Пробл эндокринол 2005; 3: 22-32.
14. Pasquier F., Boulogne A., Leys D., Fontaine P Diabetes mellitus and dementia. Diabetes Metab 2006; 32 (5 Pt 1): 403-414.
15. Ott A., Stolk R.P., van Harskamp F. et al. Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology 1999; 53 (9): 1937-1942.
16. Бондарева В.М., Чистякова О.В. Инсулин и инсулинрецепторная сигнальная система мозга. Нейрохимия Академиздатцентр «Наука» РАН 2007; 24: 8-20.
17. Gasparini L, Netzer W.J., Greengard P., Xu H. Does insulin dysfunction play a role in Alzheimers disease? Trends Pharmacol Sci 2002; 23 (6): 288-393.
18. Areosa S.A., Grimley E. V. Effect of treatment of type II diabetes mellitus on the development of cognitive impairment and demencia. Cochrain Database Syst Rev 2002; 4: CD003804.
19. Partanen J., Niskanen L., Lehtinen J. et al. Natural history of peripheral neuropathy in patients with non-insulin-dependent diabetes mellitus. N Engl J Med 333; 1995:89 —94.
20. Young M.J., Boulton A.J., MacLeod A.F et al. A multicentre study of the prevalence of diabetic peripheral neuropathy in the United Kingdom hospital clinic population. Diabetologia 1993; 36: 150— 154.
21. Veves A., Backonja M., Malik R.A. Painful diabetic neuropathy: epidemiology, natural history, early diagnosis, and treatment options. Pain Med 2008; 9 (6): 660—674.
22. Румянцева С.А., Ступин В.А., Оганов Р.Г. и др. Теория и практика лечения больных с сосудистой коморбидностью. Клиническое руководство. М—СПб: Медицинская книга 2013; 360.
23. Гусев Е.И., Скворцова В.И. Ишемия головного мозга. М: Медицина 2001; 328.
24. Шишкова В.Н. Конгнитивные нарушения как универсальный клинический синдром в практике терапевта. Тер арх 2014; 11: 128—134.
25. Занозина О.В., Боровков Н.Н, Балаболкин М.И. Необходимость и достаточность использования антиоксидантов в терапии больных сахарным диабетом 2-го типа. Бюл экспер биол 2006; приложение 1.
26. Мазуров В.И., Болотова М.Е. Роль и место Мексидола в лечении метаболического синдрома. Рус мед журн 2008; 15 (325): 1024—1028.
27. Танашян М.М. Лагода О.В., Антонова К.В. Хронические цереброваскулярные заболевания на фоне метаболического синдрома: новые подходы к лечению. Журн неврол и психиатр 2012; 11: 21—26.
28. Местер Н.В. Влияние производных 3-оксипиридина на когнитивные функции и аффективный статус больных сахарным диабетом: Автореф. дис. ... канд. мед. наук. Челябинск 2007.
29. Волчегорский И.А., Москвичева М.Г., Чащина Е.Н. Эффективность производных 3-оксипиридина и янтарной кислоты у больных сахарным диабетом с синдромом диабетической стопы. Клин мед 2004; 11: 31—35.
30. Волчегорский И.А., Москвичева И.А. Влияние препарата мексидол на проявления дистальной симметричной полиневропатии у больных сахарным диабетом с синдромом диабетической стопы. Фарматека 2007; 20 (154): 76—79.



