Аспекты муковисцидоза у детей
СтатьиО.В. Сударева, М.В. Краснов, д-р мед. наук, профессор, Т.В. Костякова
ФГБОУ ВО «Чувашский государственный университет имени И.Н. Ульянова», г. Чебоксары
Ключевые слова: муковисцидоз, диагностика, детский возраст, трансмембранный регулятор проводимости муковисцидоза, полиорганное поражение
Keywords: cystic fibrosis, diagnosis, childhood, transmembrane conductance regulator of cystic fibrosis, multiple organ damage
Резюме. Кистозный фиброз (муковисцидоз, МВ) - аутосомно-рецессивное моногенное наследственное заболевание, характеризующееся поражением всех экзокринных желез, а также жизненно важных органов и систем, обусловленное мутациями в гене трансмембранного регулятора проводимости муковисцидоза (CFTR), который располагается в середине длинного плеча 7-й хромосомы, содержащий 27 экзонов и охватывающий 250 000 пар нуклеотидов. Клиническая картина характеризуется мультисистемным повреждением органов и систем организма и желез внешней секреции. Цель исследования - обобщение и систематизация имеющихся литературных данных о причинах, роли этиопатогенетических механизмов, критериях диагностики, клинических проявлений и эффективности современных методов терапии муковисцидоза у детей. Методы. Был проведен обзор литературных источников научно-исследовательских трудов, написанных зарубежными и отечественными авторами, с применением поисковых систем, таких как PubMed и eLIBRARY. В данном анализе публикаций включены статьи, которые содержат только доказательно-экспериментальную и клиническую базу по наиболее актуальным и современным вопросам, касающимся эпидемиологии, этиологии, патогенеза, терапии МВ. Анализировались только полнотекстовые источники, находящиеся в открытом доступе. Результаты. Муковисцидоз - это мультисистемное заболевание, которое поражает дыхательную систему, поджелудочную железу, желудочно-кишечный тракт, печень, слюнные и потовые железы и репродуктивную систему. Варьирование частоты МВ зависит от этнической принадлежности или географической зоны. МВ вызывается мутациями в гене трансмембранного регулятора проводимости (CFTR). Ген CFTR образует анионный канал, транспортирующий Cl- или НСО3- и способствующий регуляции всасывания и секреции соли и воды в различных тканях. При определенных мутациях гена CFTR образуются дефектные белки CFTR, приводящие к нарушению транспорта через эпителиальные клетки и в итоге происходит закупорка слизистых выделений в протоках желез. Диагностику при муковисцидозе проводят в пренатальном и постнатальном периоде. Клиническая симптоматика муковисцидоза многообразна и характеризуется полиорганностью поражения и большим количеством вариантов мутаций гена CFTR, при котором поражаются органы-мишени. Поражение респираторной системы преобладает в клинической симптоматике муковисцидоза. Вследствие снижения локального и системного иммунитета присоединяется различная патогенная флора, поддерживающая хронический гнойно-воспалительный процесс, что требует постоянной антибактериальной терапии. В дыхательных путях наблюдается скопление слизи, нарушение мукоцилиарного клиренса, хроническое воспаление и рецидивирующие инфекции. Терапия МВ заключается в комплексном лечении с использованием антибактериальных, муколитических и бронхолитических препаратов в сочетании с ферментотерапией, витаминотерапией и с кинезитерапией. Заключение. Таким образом, проведенный обзор научных публикаций показал, что клинические проявления муковисцидоза характеризуются поражением желез внешней секреции, тяжелыми нарушениями функций органов дыхания, мультисистемным повреждением органов и систем организма, формированием хронического воспаления и рецидивирующих инфекций бактериальной и грибковой этиологии. Наиболее перспективной является дальнейшая разработка таргетной терапии, направленной на исправление функции дефектного гена трансмембранного регулятора проводимости (CFTR), позволяющей в результате воздействия на этиопатогенетические механизмы заболевания уменьшить частоту и тяжесть бронхолегочных обострений, улучшить результаты лечения.
Summary. Cystic fibrosis (cystic fibrosis, CF) is an autosomal recessive monogenic hereditary disease characterized by damage to all exocrine glands, as well as vital organs and systems, caused by mutations in the gene of the transmembrane conduction regulator of cystic fibrosis (CFTR), which is located in the middle of the long arm of chromosome 7, containing 27 exons and covering 250,000 pairs of nucleotides. The clinical picture is characterized by multisystem damage to organs and systems of the body and exocrine glands. Objective. The purpose of the study is to summarize and systematize the available literature data on the causes, role of etiopathogenetic mechanisms, diagnostic criteria, clinical manifestations and effectiveness of modern methods of treating cystic fibrosis in children. Methods. A review of the literature sources of research papers written by foreign and domestic authors was conducted using search engines such as PubMed and eLIBRARY. This analysis of publications includes articles that contain only evidence-based experimental and clinical base on the most pressing and modern issues related to the epidemiology, etiology, pathogenesis, and therapy of CF. Only full-text sources that are in the public domain were analyzed. Results. Cystic fibrosis is a multisystem disease that affects the respiratory system, pancreas, gastrointestinal tract, liver, salivary and sweat glands, and reproductive system. The incidence of CF varies by ethnicity or geographic area. CF is caused by mutations in the transmembrane conductance regulator (CFTR) gene. The CFTR gene forms an anion channel that transports Cl- or НСОЗ- and helps regulate the absorption and secretion of salt and water in various tissues. With certain mutations of the CFTR gene, defective CFTR proteins are formed, leading to impaired transport through epithelial cells and, as a result, blockage of mucous secretions in the gland ducts. Diagnosis of cystic fibrosis is carried out in the prenatal and postnatal period. The clinical symptoms of cystic fibrosis are diverse and are characterized by multiple organ involvement and a large number of variants of CFTR gene mutations that affect target organs. Damage to the respiratory system predominates in the clinical symptoms of cystic fibrosis. Due to a decrease in local and systemic immunity, various pathogenic flora are attached, which supports a chronic purulent-inflammatory process, which requires constant antibacterial therapy. The airways experience mucus accumulation, impaired mucociliary clearance, chronic inflammation, and recurrent infections. CF therapy consists of complex treatment using antibacterial, mucolytic and bronchodilator drugs in combination with enzyme therapy, vitamin therapy and kinesitherapy. Conclusion. Thus, a review of scientific publications showed that the clinical manifestations of cystic fibrosis are characterized by damage to the exocrine glands, severe dysfunction of the respiratory system, multisystem damage to organs and systems of the body, the formation of chronic inflammation and recurrent infections of bacterial and fungal etiology. The most promising is the further development of targeted therapy aimed at correcting the function of the defective transmembrane conductance regulator (CFTR) gene, which, as a result of influencing the etiopathogenetic mechanisms of the disease, can reduce the frequency and severity of bronchopulmonary exacerbations and improve treatment results.
Для цитирования: Сударева О.В., Краснов М.В., Костякова Т.В. Аспекты муковисцидоза у детей // Практика педиатра. 2025. № 1. С. 31-39.
For citation: Sudareva O.V., Krasnov M.V., Kostyakova T.V. Aspects of cystic fibrosis in children // Pediatrician's Practice. 2025;(1): 31-39. (In Russ.)
Введение
Кистозный фиброз или муковисцидоз (МКБ-10 E84.0-84.9) [1, 2] представляет собой моногенное аутосомно-рецессивное наследственное заболевание [3-5], вызываемое патологическими мутациями гена трансмембранного регулятора проводимости (CFTR) и проявляющееся полиорганным множественным поражением желез внешней секреции [6-8], а также нарушениями функционирования таких систем, как дыхательная, пищеварительная, сердечно-сосудистая и репродуктивная [1, 9].
Дисфункция белка CFTR [10, 11] вызывает изменение ионного транспорта в апикальной мембране эпителиальных клеток различных органов. Эта дисфункция начинает проявляться внутриутробно и в периоде новорожденности. Это сложное и полиморфное заболевание, классический фенотип - прогрессирующая обструктивная болезнь легких, внешнесекреторная недостаточность поджелудочной железы и повышенные уровни хлоридов и натрия в поте у 90% пациентов [12].
Актуальность изучения МВ заключается в необходимости проведения ранних диагностических мероприятий и персонализированной терапии для улучшения качества и продолжительности жизни пациентов [13]. Научный интерес к данной тематике состоит в определении взаимосвязи клинических форм, характера нарушения функций отдельных органов и систем, особенностей течения муковисцидоза с типом мутации патологического гена, а также применение современной таргетной терапии с целью совершенствования профилактических и лечебных мероприятий [14].
Цель исследования
Провести изучение и систематизацию этиопатогенетических механизмов, критериев диагностики, клинических проявлений и эффективности основных методов терапии муковисцидоза в детском возрасте.
Методы
Методом системного анализа представлен обзор научно-исследовательских трудов зарубежных и отечественных авторов с задействованием поисковых ресурсов, таких как PubMed, содержащий базу данных медицинской литературы Medline (www.pubmed. ncbi.nlm.nih.gov) и научная электронная библиотека eLIBRARY (www.elibrary.ru). В данном литературном обзоре представлены научные публикации, которые содержат доказательно экспериментальную и клиническую базу по наиболее значимым актуальным вопросам МВ. Критериями включения являлись только полнотекстовые источники, находящиеся в открытом доступе и написанные на русском и английском языках. Критериями невключения являлось многократное дублирование статей по тематике «муковисцидоз в детском возрасте» и учитывались критерии исключения: отсутствие открытого полного доступа к текстам научных публикаций. В статью были включены данные из клинических рекомендаций и актуальных научных статей по теме обзора.
Результаты исследования
Эпидемиология
Муковисцидоз распространен повсеместно и наблюдается во всех странах мира, но отличается популяционная частота выявляемости со снижением градиента с запада на восток и с севера на юг Евразии. МВ встречается с одинаковой частотой у лиц обоего пола [14].
Частота МВ может варьироваться в зависимости от этнической принадлежности или географической зоны. Выявлено, что дети с МВ могут родиться в здоровых семьях и, следовательно, выявление гетерозиготного носительства является наиболее актуальной задачей современного здравоохранения [14].
По данным национального регистра за 2011-2021 гг., число пациентов с МВ, включенных в регистр, увеличилось с 1026 (2011) до 3969 (2021). Средний возраст больных с МВ в Российской Федерации в 2021 г. составил 14,0 ± 9,8 лет, медиана возраста - 11,9 (6,7-19,0) лет [15].
По рисунку 1 видно, что в Центральном и Приволжском федеральных округах количество детей, выявленных с МВ, больше, чем в остальных. В основном МВ рассматривается как детское заболевание, но в последние десятилетия количество взрослых выросло, по данным национального регистра, с 25 до 27,4% (2021) [15]. Реже по количеству выявленных случаев данная патология встречается у жителей Африки и Японии [16].

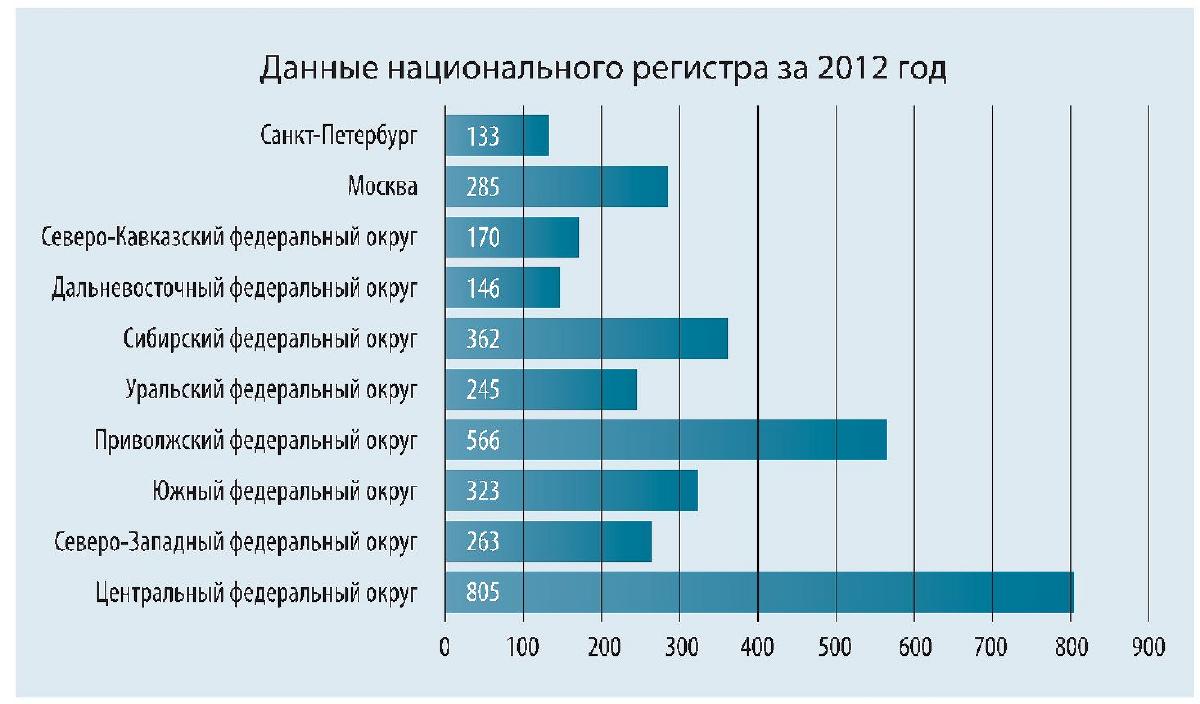
Рис.1. Абсолютное число детей с МВ в каждом округе РФ
Этиология и патогенез
При муковисцидозе происходят мутации в гене трансмембранного регулятора проводимости (CFTR), расположенного на длинном плече 7-й хромосомы в участке 7q31.2 и способного кодировать белок CFTR, что приводит к нарушению функционирования белка CFTR на поверхности клетки [12, 17-19].
Трансмембранный регулятор проводимости муковисцидоза отвечает за транспорт ионов хлора через клетки апикального эпителия в тканях дыхательных путей, кишечника, поджелудочной железы, почек, потовых желез и мужского репродуктивного тракта. Также он выполняет множество других регуляторных функций, таких как секреция бикарбоната, регулирующего рН жидкости на поверхности дыхательных путей, и ингибирование эпителиального натриевого канала, играющего важную роль в гидратации секрета и муцинов [20].
CFTR относится к семейству белков ABC-транспортеров (АТФ-связывающих кассет), разделяющих трансмембранные транспортные функции и включает около 250 000 пар нуклеотидов геномной последовательности и состоящий из 27 экзонов. Белок CFTR образует анионный канал, транспортирующий Cl- и НСО3-, функционирование которого регулируется фосфорилированием, опосредованным цАМФ-зависимыми фосфокиназами. При возникновении мутации в гене CFTR могут формироваться дефектные белки и несколько мутировавших белковых молекул CFTR, которые способны достигать клеточной мембраны и являются дисфункциональными и в результате не могут осуществлять транспорт хлорид-ионов, что способствует накоплению хлорид-ионов и связанных с ними молекул воды в эпителиальных клетках и отсутствию гидратации внеклеточной слизи и секрета [21].
Белок CFTR включает 1480 аминокислот одной полипептидной цепи, которая имеет 5 доменов: два нуклеотид-связывающих домена (NBD), два трансмембранных домена (MSD) и регуляторный (R) домен [22, 23].
Согласно данным национального регистра, наиболее часто встречаются следующие мутации: F508del (53,14%), CFTRdele2,3 (6,18%), E92K (3,11%), 3849+10kbC>T (2,29%), 2143delT (2,05%), 2184insA (1,88%), 1677delTA (1,76%), N1303K (1,69%), W1282X (1,63%), L138ins (1,46%), G542X (1,43%) [2, 15]. У европеоидов североевропейского происхождения наиболее распространенной патогенной мутацией является мутант делеции, обозначенный F508del (с делецией фенилаланина в участке 508, вызванной геномной делецией трех нуклеотидов, обозначенных c.1521_1523delCTT) [21].
Различные мутации CFTR могут нарушать синтез, обработку, стабильность плазматической мембраны, рециркуляцию или стробирование белка CFTR с различными фенотипическими последствиями в зависимости от количества остаточного функционального белка, таким образом мутации CFTR [7] начали классифицировать в различные классы [7, 24]. По данным национального консенсуса (2-е издание) (2018) по МВ, в зависимости от влияния на функцию белка CFTR все варианты нуклеотидной последовательности гена CFTR подразделяют на семь основных классов: класс I - нарушение синтеза белка (нарушение транскрипции и трансляции, приводящие к нестабильному, усеченному или отсутствующему белку); II класс - нарушение созревания белка (дефекты созревания белка и транспортировки к мембране); III класс - нарушение регуляции хлорного канала (дефекты регулирования отверстия канала в апикальной мембране); IV класс - нарушение проводимости хлорного канала (миссенс-мутации, располагающиеся в мембраносвязанных доменах, которые приводят к дефектам потока ионов по каналу, то есть изменение проводимости хлорного канала вследствие сокращения времени открытия ионного канала); класс V - снижение количества функционального белка (продуцируется пониженное количество нормального транскрипта, или снижается уровень функционального белка, или понижен уровень транспорта молекул белка CFTR, в результате которых нарушается механизм сплайсинга); класс VI -снижение времени нахождения белка на поверхности клетки (синтез протеина с измененной стабильностью в результате потери 70-98 С-концевых аминокислотных остатков). Также выделяют VII класс, при котором нарушается образование иРНК, вследствие перестройки гена CFTR (обширные перестройки гена CFTR (делеции, инсерции), нормальный сплайсинг либо мутации, изменяющие донорный или акцепторный сайты сплайсинга одного экзона) [1, 25]. Варианты I-III и VII классов обычно вызывают тяжелую форму муковисцидоза, в то время как мутации классов IV-VI вызывают более легкую его форму [26].
В развитии патогенеза муковисцидоза выделяют три основных механизма: поражение соединительной ткани и экзокринных желез, нарушение электролитного обмена. Проявление патологии экзокринных желез сопровождается продукцией более вязкого секрета с повышенным содержанием белка и некоторых электролитов и снижением активности ферментов. Основой бронхолегочных изменений служит нарушение проходимости и процесса самоочищения бронхов [16].
Воспаление играет важнейшую роль в муковисцидозе и в результате в прогрессировании заболевания [19, 27]. Литературные данные подтверждают, что воспалительный процесс у пациентов с муковисцидозом чаще всего характеризуется иммунным ответом 2-го типа [19].
Таким образом, в связи с мутацией в гене регулятора трансмембранной проводимости муковисцидоза, приводящего к дисфункциональному белку CFTR, ухудшается транспортировка хлорид-ионов к поверхности клеток, что приводит к накоплению вязкого секрета в легких, поджелудочной железе, кишечнике и гепатобилиарных протоках, в результате чего возникает обструкция и фиброз [28]. В поджелудочной железе заболевание вызывает обструкцию протоков железы, появление кист, внутреннюю и внешнесекреторную недостаточность и кишечную мальабсорбцию. При муковисцидозе в печени формируется холестаз, холелитиаз и вторичный цирроз печени [25].
При разных типах патологических мутаций происходит выработка разного количество белка или белок CFTR может иметь различия по функциональным характеристикам, что отражается на клинической картине МВ [13].
Также в бронхолегочной системе происходит развитие хронической колонизации, что является важной особенностью патогенеза заболевания. Происходит заселение грамположительной кокковой микробной флорой бронхолегочной системы в начале жизни ребенка с МВ, но затем начинают преобладать грамотрицательные неферментирующие бактерии, вызывающие тяжелые формы инфекционного процесса. В частности, могут способствовать хронизации патологических процессов нетуберкулезные микобактерии [16].
Диагностика
Диагностика МВ включает: пренатальную диагностику, диагностику по неонатальному скринингу (до клинических проявлений), диагностику при клинических проявлениях и диагностику у родственников больных [29].
Потребность в пренатальной диагностике наследственных генетических заболеваний существенно возрастает в связи с ростом частоты идентифицируемых генов заболеваний и совершенствованием методов диагностики. Пренатальный скрининг на муковисцидоз предлагает будущей матери и ее супругу оценить потенциальный риск аномалии их ребенка [30].
Пренатальное тестирование плода на CFTR проводят только в случае повышенного риска, когда оба родителя являются носителями одного дефектного варианта CFTR или когда при проведении УЗИ на втором триместре выявляется эхогенность кишечника плода. Пренатальное инвазивное тестирование CFTR обычно проводится путем забора ворсин хориона или амниоцентеза, однако клиническая практика в настоящее время смещается в сторону неинвазивного пренатального тестирования на МВ [31]. При проведении амниоцентеза возможна ДНК-диагностика (получение околоплодных вод на определенных сроках беременности: ранний в 13-14 недель и поздний в 16-20 недель) в семье носителей одной мутации гена CFTR и имеющей больного ребенка с МВ или в семье, где был больной ребенок с МВ [29].
Для неинвазивного пренатального теста [32, 33] для диагностики муковисцидоза используют либо бесклеточную ДНК плода, либо циркулирующие трофобласты в материнской крови. Использование бесклеточной ДНК плода изначально было основано на обнаружении или исключении аллеля отцовского варианта в материнской плазме. Этот подход полезен только в том случае, если родители являются носителями разных вариантов муковисцидоза, и поэтому не применим для скрининга муковисцидоза в популяции с высокой однородностью мутаций. Кроме того, обнаружение аллеля отцовского варианта потребует инвазивного тестирования для определения состояния плода для аллеля материнского варианта. Вместо этого была разработана стратегия косвенного тестирования, основанная на анализе относительной дозировки гаплотипа [32].
Скрининг новорожденных (NBS) на муковисцидоз [34, 35] был впервые введен в 1980-х гг. и проводится путем измерения уровня иммунореактивного трипсиногена (IRT) при скрининге пятен крови новорожденных. У новорожденных с муковисцидозом слизистые пробки частично блокируют протоки поджелудочной железы, ведущие в тонкий кишечник, не давая трипсиногену попасть в кишечник и преобразоваться в панкреатический фермент поджелудочной железы трипсин. Скрининг считается положительным, если уровень IRT остается постоянно повышенным в возрасте от 7 до 14 дней жизни или если при генетическом тестировании выявлен хотя бы один из вариантов CFTR [35].
Протокол скрининга в РФ включает три обязательных этапа: IRT, ретест IRT и потовый тест. На первом этапе в крови новорожденных (1-2-й день - у доношенных, 7-8-й день - у недоношенных) определяется уровень IRT. При превышении порогового уровня ИРТ проводится ретест на 21-28-й день жизни [25].
В РФ используются две методики потового теста: 1) классический прямой метод определения электролитного состава пота (хлора или натрия) метод Гибсона - Кука (1959 г.), который является стандартным методом, рекомендован национальными и международными рекомендациями и основан на выявлении повышенных значений хлорида пота с помощью количественного теста ионофореза пилокарпина (QPIT). Младенец с нормальной концентрацией хлорида пота <30 ммоль/л и только одной мутацией считается носителем, и при определении значений тестирования хлорида пота в двух независимых измерениях с показателем >60 ммоль/л или по результатам теста, когда в последовательности гена CFTR выявлены два вызывающих заболевание варианта муковисцидоза, только тогда диагноз МВ подтверждается [1, 36]; 2) потовая проба проводится путем определения проводимости пота с помощью специальных потовых анализаторов, коррелируя с определением уровня хлоридов и позволяя получить результат при количестве пота в образце 3-10 мкл и положительными считаются результаты >80 ммоль/л, показатели 50-80 ммоль/л -пограничными, менее <50 ммоль/л - отрицательными. Данный метод определения проводимости пота получил широкое распространение при внедрении массового скрининга новорожденных [1, 25] и во многих российских центрах используется система «Macroduct» в комплексе с потовым анализатором «Sweat-Chek» (ELITechGroup Inc., США), но для обследования новорожденных разработана и применяется система «Nanoduct» (Wescor, США), для которой необходимо минимальное количество потовой жидкости (3-6 мкл) [1, 25, 36].
Анализ вариаций генов CFTR определяют расширенным ДНК-анализом (секвенирование гена): высокопроизводительным секвенированием (секвенирование следующего поколения, NGS) для обнаружения мутаций и секвенированием по Сэнгеру [37]. Секвенированием можно выявить нарушения последовательности гена, незначительные по протяженности: нуклеотидные замены, небольшие делеции/инсерции. Перестройки, охватывающие несколько экзо-нов/интронов, такими методами не выявляются. Рекомендуется использовать следующие технологии: MLPA - мультиплексную лигазную зондовую амплификацию либо QFMP - количественную флуоресцентную мультиплексную ПЦР [1].
Диагноз заболевания МВ [38] подтверждается при наличии положительного неонатального скрининга, положительного потового теста и ДНК-диагностики с определением двух мутаций CFTR, вызывающих МВ (согласно базе CFTR-2 cftr2.org) [1].
Клинические проявления
Белок CFTR экспрессируется во многих тканях, с высокой экспрессией в эпителии дыхательных путей, тонкой и толстой кишки, панкреатических и гепатобилиарных протоков, мужских репродуктивных путей, что приводит к прогрессирующей дисфункции и повреждению органов, что обусловливается полиорганностью поражения в клинических проявлениях [39].
Всемирная организация здравоохранения, Международная ассоциация муковисцидоза, Европейская ассоциация муковисцидоза используют в настоящее время классификацию по МКБ-10, кистозный фиброз (E84): E84.0 кистозный фиброз с легочными проявлениями; E84.1 кистозный фиброз с кишечными проявлениями; E84.8 кистозный фиброз с другими проявлениями; E84.9 кистозный фиброз неуточненный [1, 25].
По данным клинической классификации, составленной на основе рабочей классификации С.В. Рачинского, рекомендаций ВОЗ и Европейской ассоциации муковисцидоза, выделяют классический МВ, смешанную или легочно-кишечную форму заболевания и легочную форму заболевания [1, 25, 40].
Муковисцидоз поражает несколько органов-мишеней, при этом в клинической картине преобладают персистирующая бактериальная инфекция и хроническое нейтрофильное воспаление в дыхательных путях [41]. В дыхательных путях нарушение функции этого белка приводит к увеличению густоты слизи, которая не очищается мукоцилиарной системой, что способствует хронизации инфекции в дыхательных путях и дальнейшему неконтролируемому воспалительному процессу [42]. При классической и смешанных формах МВ клиническая картина в основном представлена хроническим обструктивным бронхитом и бронхоэктазами, а легочная форма в свою очередь - пневмофиброзом. Данные формы также могут сопровождаться другими проявлениями заболевания, такими как синуситы, синдромом псевдо-Барттера, азооспермией и рецидивирующим панкреатитом [1, 25].
В клинике МВ бронхолегочные патологические изменения преобладают и определяют прогнозирование и вследствие тяжесть заболевания у 95% больных. У детей в очень раннем возрасте развивается симптоматика повреждения органов дыхания: присутствие постоянного или стойкого сухого кашля, обычно усиливающегося в ночное время или после пробуждения; возникновение удушья, одышки и временами рвоты [3]. При измененной реологии секрета дыхательных путей его клиренс затрудняется с развитием хронической обструкции мелких дыхательных путей, хронической инфекции патогенными микроорганизмами и бронхоэктазов с деструкцией тканей [43, 44].
Течение клиники заболевания осложняется частыми периодами острого ухудшения состояния легких, называемого «легочным обострением» [44, 45].
Основной причиной заболеваемости и смертности при МВ является прогрессирующее заболевание легких, обусловленное сложным и разнообразным воспалительным иммунным синдромом, вызванным рецидивирующими и хроническими бактериальными и грибковыми инфекциями легких [46]. Другими распространенными клиническими проявлениями при МВ являются задержка роста, недостаточность поджелудочной железы, мекониальная непроходимость и бесплодие. Таким образом, при муковисцидозе в основном поражаются легкие и поджелудочная железа, а также верхние дыхательные пути, печень, кишечник и репродуктивные органы. 99% пострадавших пациентов мужского пола бесплодны из-за обструктивной азооспермии, а 87% пациентов имеют экзокринную недостаточность поджелудочной железы. Данное заболевание обусловлено тем, что сокращается продолжительность жизни, а медиана прогнозируемого возраста выживания и медиана ожидаемой продолжительности жизни составляют 47,4 и 44,4 года соответственно [47]. Желудочно-кишечные симптомы также связаны с экзокринной недостаточностью поджелудочной железы, которая нарушает процессы пищеварения и всасывания, что вызывает хроническую стеаторею, боль в желудке и, как следствие, недостаточную массу тела и рост. Реже наблюдаются изменения в печени и желчных протоках [48]. Таким образом, типичные клинические проявления локализуются в дыхательном и желудочно-кишечном трактах [49].
Поражение гепатобилиарной системы обычно проявляется в конце первого десятилетия жизни, то есть в младшем школьном возрасте, и имеет распространенность в диапазоне 5,7-39%. Патология является третьей причиной летального исхода у пациентов с муковисцидозом, которому предшествует только поражение легких и осложнения, присущие трансплантации. Кроме того, некоторые исследования показали, что пациенты со значительным повреждением печени имеют более высокий риск ухудшения дополнительных внепеченочных проявлений, такие как снижение веса, сахарный диабет, печеночная остеодистрофия и легочные заболевания, что приводит к обострению заболеваемости в основное заболевание. По этой причине гепатобилиарное заболевание было признано фактором риска неблагоприятного развития и неблагоприятного прогноза [49]. Изменения в печени в ходе муковисцидоза представляют собой сеть сложных процессов фиброза, воспаления, ремоделирования, апоптоза и холестаза, возникающих в результате аномального функционирования белка CFTR, иммунологических реакций и реакции на окислительный стресс. Наиболее распространенные изменения, наблюдаемые в печени и желчных протоках, включают очаговый фиброз, стеатоз печени, билиарный цирроз печени, портальную гипертензию и/или холедохолитиаз. У большинства пациентов с муковисцидозом течение печеночных осложнений начинается бессимптомно [48, 50, 51].
Пациенты с муковисцидозом подвержены риску развития острого повреждения почек из-за приема потенциально нефротоксичных препаратов, таких как антибиотики. Аминогликозиды являются примерами препаратов, которые участвуют в повреждении почек, что связанно с повреждением канальцев. Повреждение канальцев в основном затрагивает эпителиальные клетки проксимальных отделов, особенно из-за их метаболической нагрузки и участия в секреции и реабсорбции клубочковых фильтратов, что подвергает их воздействию нефротоксинов [52].
Лечение
Стандартная обязательная терапия МВ в России включает: диетотерапию, заместительную терапию недостаточности экзокринной функции поджелудочной железы, муколитическую терапию, дренирование бронхиального дерева и лечебную физкультуру, антибактериальную терапию, витаминотерапию, предупреждение и лечение обострений инфекционно-воспалительного процесса в околоносовых пазухах для уменьшения степени нисходящей контаминации бронхолегочной системы и терапию осложнений [1].
На причину возникновения МВ, а именно на работу белка CFTR, эти препараты не воздействуют, что способствовало разработке новых медикаментов, влияющих на патогенез МВ [53], так называемых модуляторов, включающих такие препараты, как корректоры и потенциаторы CFTR [53-55].
Потенциаторы и корректоры одобрены Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США (Foodand Drug Administration; FDA), и они применяются в лечении МВ по настоящее время [56]. Корректоры способствуют тому, что мутантный белок CFTR при прохождении через систему внутриклеточного качественного контроля занимает правильное местоположение на апикальной мембране (применяются при мутациях II класса), к ним относятся: 4-фенилбутират/генистин, аналог силденафила-КМ11060, куркумин, VX-809, VX-661. А потенциаторы воздействуют на молекулы мутантного белка CFTR, расположенные в апикальной мембране и таким образом активизируют или восстанавливают функцию ионного канала (мутации III-IV классов), к ним относится генистин (VX-770) [54].
Для лечения пациентов применяются четыре модулятора CFTR, нацеленные на дефект белка CFTR: корректоры лумакафтор, тезакафтор и элексакафтор и потенциатор ивакафтор. Комбинация одного корректора и потенциатора приводит к умеренному клиническому эффекту у пациентов, гомозиготных по F508del, в то время как комбинация тезакафтора, элексакафтора и ивакафтора улучшает исход у пациентов с одной или двумя мутациями F508del [57].
Первым модулятором для лечения гомозигот по F508del при МВ стал комбинированный препарат, который состоит из ивакафтора (VX-770) и лумакафтора (VX-809). В России данный препарат был зарегистрирован в декабре 2020 г. Эффективность и безопасность Ива/Лум у пациентов в возрасте >12 лет с объемом форсированного выдоха за первую секунду (ОФВ1) от 40 до 90% от должного была изучена в двух 24-недельных плацебо-контролируемых в параллельных группах исследованиях фазы III (исследование TRAFFIC и TRANSPORT) [53]. Рекомендуется применение комбинации ивакафтор + лумакафтор (данный препарат разрешен в РФ с 2 лет согласно инструкции) у пациентов с МВ гомозиготных по мутации F508del в гене CFTR с целью патогенетического лечения, повышения количества активного белка CFTR на поверхности клеток экзокринных желез, повышения легочной функции, снижения частоты легочных обострений и замедления прогрессирования заболевания [58].
Лечение потенциатором ивакафтором приносит большую пользу пациентам с мутацией III класса и умеренную пользу пациентам с выбранным списком мутаций остаточной функции, облегчая перенос хлора и повышая способность открытия канала (или гейтинг) белка CFTR на поверхности клетки. Данный препарат показан к применению у детей с 4 месяцев и старше [57, 58].
Препараты элексакафтор и тезакафтор показаны с 6-летнего возраста и старше и действуют как корректоры CFTR, восстанавливая процессинг F508del, связываясь с белком CFTR, чтобы увеличить доступность белка CFTR на клеточной поверхности. Они работают, изменяя форму белка CFTR для его расположения на поверхности клетки [58].
Для подавления инфекционного процесса в верхних дыхательных путях, бронхах и легких назначаются антибиотики β-лактамного ряда (цефалоспорины 4-го поколения, имипенем, азлоциллин, пиперациллин карбапенемы и пенициллины), фторхинолоны (офлоксацин и ципрофлоксацин и др.) и аминогликозиды (тобрамицин и гентамицин). Для проведения санации от вязкого секрета полости носа, околоносовых пазух и бронхов применяют ингаляции муколитиков: дорна-за-альфа, ацетилцистеин в комбинации с туаминогептаном и физиотерапевтические методики лечения [13].
Анализ проведенных за последнее десятилетие исследований показал [59], что лечение пациентов с муковисцидозом должно подбираться индивидуально для каждого пациента и также быть комплексным и постоянным [13].
Заключение
Таким образом, проведенный обзор научных публикаций показал, что клинические проявления муковисцидоза характеризуются поражением желез внешней секреции, тяжелыми нарушениями функций органов дыхания, мультисистемным повреждением органов и систем организма, формированием хронического воспаления и рецидивирующих инфекций бактериальной и грибковой этиологии. Наиболее перспективной является дальнейшая разработка таргетной терапии, направленной на исправление функции дефектного гена трансмембранного регулятора проводимости (CFTR), позволяющей в результате воздействия на этиопатогенетические механизмы заболевания уменьшить частоту и тяжесть бронхолегочных обострений и в результате улучшить результаты лечения. Также необходимо проводить изучение распространенности муковисцидоза с целью выявления уникальных и новых генетических вариантов, разрабатывать диагностические методы исследования и изучать особенности проявлений клинической картины муковисцидоза.
Литература
1. Кистозный фиброз (муковисцидоз) : клинические рекомендации. 2021. URL: mukoviscidoz.org/doc/KP372. pdf (дата обращения: 29.08.2024).
2. Современные подходы к ведению детей с муковисцидозом / А.А. Баранов [и др.] // Педиатрическая фармакология. 2022. Т. 19, № 2. С. 153-195. DOI: 10.15690/pf.v19i2.2417.
3. Красовский С.А., Самойленко В.А., Амелина Е.Л. Муковисцидоз: диагностика, клиника, основные принципы терапии // Атмосфера. Пульмонология и аллергология. 2013. № 1. С. 42-46.
4. Поляков Д.П., Карнеева О.В., Белавина П.И. Хронический риносинусит у детей с муковисцидозом: современные тенденции диагностики и лечения // Российская ринология. 2018. Т. 26, № 4. С. 17-25. DOI:10.17116/rosrino20182604117.
5. Debray D., Corvol H., Housset C. Modifier genes in cystic fibrosis-related liver disease // Current Opinion in Gastroenterology. 2019. Vol. 35, No. 2. P. 88-92. DOI: 10.1097/ MOG.0000000000000508.
6. Cystic fibrosis liver disease: A condition in need of structured transition and continuity of care / J. Hercun, F. Alvarez, C. Vincent, M. Bilodeau // Canadian Liver Journal. 2019. Vol. 2, No. 3. P. 71-83. DOI: 10.3138/canlivj-2018-0019.
7. Maiuri L., Raia V., Kroemer G. Strategies for the etiological therapy of cystic fibrosis // Cell Death and Differentiation. 2017. Vol. 24, No. 11. P. 1825-1844. DOI: 10.1038/cdd.2017.126.
8. Nutritional Care in Children with Cystic Fibrosis / E. Mariot-ti Zani [et al.] // Nutrients. 2023. Vol. 15, No. 3. P. 479. DOI: 10.3390/nu15030479.
9. Оценка нутритивного статуса у детей с муковисцидозом / К.О. Логвиненко [и др.] // Молодежный инновационный вестник. 2018. Т. 7, № S1. С. 93-94.
10. Mainz J.G., Koitschev A. Management of chronic rhinosinusitis in CF // Journal of Cystic Fibrosis. 2009. Vol. 8 (S1). P. 10-4. DOI: 10.1016/S1569-1993(09)60005-9.
11. Clinical Consequences and Functional Impact of the Rare S737F CFTR Variant and Its Responsiveness to CFTR Modulators / V. Terlizzi [et al.] // International Journal of Molecular Sciences. 2023. Vol. 24, No. 7. P. 6576. DOI: 10.3390/ijms24076576.
12. Cystic fibrosis: current concepts / J.A. Lopez-Valdez [et al.] // Boletrn Medico del Hospital Infantil de Mexico. 2021. Vol. 78, No. 6. P. 584-596. DOI: 10.24875/BMHIM.20000372.
13. Муковисцидоз - актуальная проблема медицины / Э.М. Эседов [и др.] // Вестник оториноларингологии. 2016. Т. 81, № 5. С. 15-18. DOI: 10.17116/otorino201681515-18.
14. Аюпова Г.Р., Минниахметов И.Р., Хусаинова Р.И. Проблемы и достижения в изучении клинико-генетических аспектов муковисцидоза // Казанский медицинский журнал. 2022. Т. 103, № 4. С. 628-640. DOI: 10.17816/KMJ2022-628.
15. Регистр пациентов с муковисцидозом в Российской Федерации. 2021 год / под ред. С.А. Красовского, М.А. Стариновой, А.Ю. Воронковой и др. М. : ИД «Медпрактика-М», 2023. URL: ostrovaru.com/wp-content/uploads/2023/10 registr-2021.pdf (дата обращения: 29.08.2024).
16. Кистозный фиброз (муковисцидоз) / С.С. Яшин, Ю.Р. Юнусова, Н.В. Исакова, Ю.В. Сердобольская // Современные проблемы науки и образования. 2022. № 5. С. 141. DOI: doi.org/10.17513/spno.32008. URL: science-edu-cation.ru/article/view (дата обращения: 05.03.2024).
17. Узденов М.Б., Узденова К.А. Генетические особенности муковисцидоза у детей в Карачаево-Черкесской Республике // Вестник молодого ученого. 2020. Т. 9, № 3. С. 89-92.
18. Phenotypic spectrum and genetic heterogeneity of cystic fibrosis in Sri Lanka / N.L.R. Indika [et al.] // BMC Medical Genetics. 2019. Vol. 20, No. 1. P. 89. DOI: 10.1186/s12881-019-0815-x.
19. Type 2 inflammation in cystic fibrosis: new insights / S. Manti [et al.] // Pediatric Allergy and Immunology. 2022. Vol. 33, No. 27. P. 15-17. DOI: 10.1111/pai.13619.
20. Dornase alfa in Cystic Fibrosis: indications, comparative studies and effects on lung clearance index / V. Terlizzi, C. Castellani, G. Taccetti, B. Ferrari // Italian Journal of Pediatrics. 2022. Vol. 48, No. 1. P. 141. DOI: 10.1186/s13052-022-01331-5.
21. Chen Q., Shen Y., Zheng J. A review of cystic fibrosis: Basic and clinical aspects // Animal Models and Experimental Medicine. 2021. Vol. 4, No. 3. P. 220-232. DOI: 10.1002/ame2.12180.
22. Link S.L., Nayak R.P. Review of Rapid Advances in Cystic Fibrosis // Missouri Medicine. 2020. Vol. 117, No. 6. P. 548-554.
23. Research advances in molecular mechanisms underlying the pathogenesis of cystic fibrosis: From technical improvement to clinical applications (Review) / T. Wei [et al.] // Molecular Medicine Reports. 2020. Vol. 22, No. 6. P. 4992-5002. DOI: 10.3892/mmr.2020.11607.
24. Pediatric population with cystic fibrosis in the centre of Portugal: candidates for new therapies / J. Roda [et al.] // Jornal de Pediatria (Rio J). 2022. Vol. 98, No. 2. P. 212-217. DOI: 10.1016/j.jped.2021.05.010.
25. Национальный консенсус Муковисцидоз: определение, диагностические критерии, терапия» / под ред. Е.И. Кондратьевой, Н.Ю. Каширской, Н.И. Капранова. 2-е изд. М. : ООО «Компания БОРГЕС», 2018.
26. A comprehensive review of cystic fibrosis in Africa and Asia / K. Abubakar Bobbo [et al.] // Saudi Journal of Biological Sciences. 2023. Vol. 30, No. 7. P. 103685. DOI: 10.1016/j.sjbs.2023.103685.
27. Roesch E.A., Nichols D.P., Chmiel J.F. Inflammation in cystic fibrosis: An update // Pediatric Pulmonology. 2018. Vol. 53, No. S3. P. S30-S50. DOI: 10.1002/ppul.24129.
28. Mason K.A., Rogol A.D. Trends in Growth and Maturation in Children with Cystic Fibrosis Throughout Nine Decades // Frontiers in endocrinology. 2022. Vol. 13. P. 935354. DOI: 10.3389/fendo.2022.935354.
29. Актуальные вопросы диагностики муковисцидоза / Е.И. Кондратьева, Н.И. Капранов, В.Д. Шерман, Н.Ю. Каширская // Практика педиатра. 2015. № 2. С. 20-27.
30. A Cross-Sectional Study of the Marital Attitudes of Pregnant Women at Risk for Cystic Fibrosis and Psychological Impact of Prenatal Screening / Z.L. Popa [et al.] // International Journal of Environmental Research and Public Health. 2022. Vol. 19, No. 14. P. 8698. DOI: 10.3390/ijerph19148698.
31. Prenatal genetic testing for cystic fibrosis: a systematic review of clinical effectiveness and an ethics review / S.J.M. Kessels [et al.] // Genetics in Medicine. 2020. Vol. 22, No. 2. P. 258-267. DOI: 10.1038/s41436-019-0641-8.
32. Noninvasive prenatal screening for cystic fibrosis using circulating trophoblasts: Detection of the 50 most common disease-causing variants / L.D. Jeppesen [et al.] // Prenatal Diagnosis. 2023. Vol. 43, No. 1. P. 3-13. DOI: 10.1002/pd.6276.
33. The Number of Circulating Fetal Extravillous Trophoblasts Varies from Gestational Week 6 to 20 / K. Ravn [et al.] // Reproductive Sciences. 2020. Vol. 27, No. 12. P. 2170-2174. DOI: 10.1007/s43032-020-00243-1.
34. Diagnostic and Communication Challenges in Cystic Fibrosis Newborn Screening / J.K. DeCelie-Germana [et al.] // Life (Basel). 2023. Vol. 13, No. 8. P. 1646. DOI: 10.3390/life13081646.
35. Dickinson K.M., Collaco J.M. Cystic Fibrosis // Pediatrics in review. 2021. Vol. 42, No. 2. P. 55-67. DOI: 10.1542/pir.2019-0212.
36. Функциональные методы диагностики нарушений гена CFTR и его продукта / Е.И. Кондратьева, Ю.Л. Мельяновская, В.Д. Шерман [и др.] // Вопросы практической педиатрии. 2018. Т. 13, № 4. С. 50-64. DOI: 10.20953/1817-7646-2018-4-50-64.
37. Clinical and gene mutation features of cystic fibrosis: an analysis of 8 cases / N. Zhang [et al.] // Zhongguo Dang Dai Er Ke Za Zhi. 2022. Vol. 24, No. 7. P. 771-777. DOI: 10.7499/j.issn.1008-8830.2203015.
38. Genetic Modifying Factors of Cystic Fibrosis Phenotype: A Challenge for Modern Medicine / L.I. Butnariu [et al.] // Journal of Clinical Medicine. 2021. Vol. 10, No. 24. P. 5821. DOI: 10.3390/ jcm10245821.
39. Cystic Fibrosis-Related Diabetes Workshop: Research Priorities Spanning Disease Pathophysiology, Diagnosis, and Outcomes / M.S. Putman [et al.] // Diabetes. 2023. Vol. 72, No. 6. P. 677-689. DOI: 10.2337/db22-0949.
40. Ларина В.С., Попова А.Е. Сочетанная форма муковисцидоза, тяжелое течение // Российский педиатрический журнал. 2022. Т. 3, № 2. С. 411-412.
41. Wang G., Nauseef W.M. Neutrophil dysfunction in the pathogenesis of cystic fibrosis // Blood. 2022. Vol. 139, No. 17. P. 26222631. DOI: 10.1182/blood.2021014699.
42. Blanchard A.C., Waters V.J. Microbiology of Cystic Fibrosis Airway Disease // Seminars in Respiratory and Critical Care Medicine. 2019. Vol. 40, No. 6. P. 727-736. DOI: 10.1055/s-0039-1698464.
43. A Novel Pathogenic Variant of the CFTR Gene in a Patient with Cystic Fibrosis Phenotype-c.4096A > T / A.B. Arslan, A.G. Zamani, S. Pekcan, M.S. Yildirim // Journal of Pediatric Genetics. 2020. Vol. 9, No. 1. P. 40-43. DOI: 10.1055/s-0039-1694964.
44. Sweat metabolomics before and after intravenous antibiotics for pulmonary exacerbation in people with cystic fibrosis / F.W. Woodley [et al.] // Respiratory Medicine. 2022. Vol. 191. P. 106687. DOI: 10.1016/j.rmed.2021.106687.
45. Evaluating FEV1 decline in diagnosis and management of pulmonary exacerbations in children with cystic fibrosis / D.C. Bouzek [et al.] // Pediatric Pulmonology. 2022. Vol. 57, No. 7. P. 17091716. DOI: 10.1002/ppul.25925.
46. Eschenhagen P., Grehn C., Schwarz C. Prospective Evaluation of Aspergillus fumigatus-Specific IgG in Patients With Cystic Fibrosis // Frontiers in Cellular and Infection Microbiology. 2021. Vol. 10. P. 602836. DOI: 10.3389/fcimb.2020.602836.
47. Current Status of Genetic Diagnosis Laboratories and Frequency of Genetic Variants Associated with Cystic Fibrosis through a Newborn-Screening Program in Turkey / S.T. Bozdogan [et al.] // Genes. 2021. Vol. 12, No. 2. P. 206. DOI: 10.3390/genes12020206.
48. Assessment of Liver Fibrosis with the Use of Elastography in Paediatric Patients with Diagnosed Cystic Fibrosis / S. Wiecek [et al.] // Disease Markers. 2022. Vol. 2022. P. 4798136. DOI: 10.1155/2022/4798136.
49. Portillo Mino J.D., Ceron Munoz E.E. Unusual presentation of CF in an infant // Respiratory Medicine Case Reports. 2020. Vol. 30. P. 101110. DOI: 10.1016/j.rmcr.2020.101110.
50. Risk factors for hepatic steatosis in adults with cystic fibrosis: Similarities to non-alcoholic fatty liver disease / F. Ayoub, C. Trillo-Alvarez, G. Morelli, J. Lascano // World Journal of Hepatology. 2018. Vol. 10, No. 1. P. 34-40. DOI: 10.4254/wjh.v10.i1.34.
51. Kamal N., Surana P., Koh C. Liver disease in patients with cystic fibrosis // Current Opinion in Gastroenterology. 2018. Vol. 34, No. 3. P. 146-151. DOI: 10.1097/MOG.0000000000000432.
52. Renal Function in Patients with Cystic Fibrosis: A Single-Center Study / M. Rachel, S. Galiniak, M. Biesiadecki, A. Gala-Bladzins ka // International Journal of Environmental Research and Public Health. 2022. Vol. 19, No. 9. P. 5454. DOI: 10.3390/ijerph19095454.
53. Каширская Н.Ю., Петрова Н.В., Зинченко Р.А. Клиническая эффективность и безопасность комбинированного препарата ивакафтор/лумакафтор у пациентов с муковисцидозом : обзор международных исследований // Вопросы современной педиатрии. 2021. Т. 20, № 6s. С. 558-566. DOI: 10.15690/vsp.v20i6S.2363.
54. Куцев С.И., Ижевская В.Л., Кондратьева Е.И. Таргетная терапия при муковисцидозе // Пульмонология. 2021. Т. 31, № 2. С. 226-236. DOI: 10.18093/0869-0189-2021-31-2-226-236.
55. Генотип-фенотипические корреляции течения кистозного фиброза у российских детей. Первое описание одиннадцати новых мутаций / Ю.В. Горинова [и др.] // Вопросы современной педиатрии. 2018. Т. 17, № 1. С. 61-69. DOI: 10.15690/vsp.vl7il.l856.
56. A review of Trikafta: triple cystic fibrosis transmembrane conductance regulator (CFTR) modulator therapy / A. Zaher [et al.] // Cureus. 2021. Vol. 13, No. 7. P e16144. DOI: 10.7759/ cureus.16144.
57. Correction of CFTR function in intestinal organoids to guide treatment of cystic fibrosis / A.S. Ramalho [et al.] // The European Respiratory Journal. 2021. Vol. 57, No. 1. P. 1902426. DOI: 10.1183/13993003.02426-2019.
58. Методические рекомендации: Таргетная терапия кистозного фиброза (муковисцидоза). 2023. URL: https:// mukoviscidoz.org/doc/med_doc/target-metod-recomend-2023. pdf (дата обращения: 29.08.2024).
59. Асманов А.И., Пивнева Н.Д. Мукоактивные препараты растительного происхождения в профилактике и терапии заболеваний ЛОР-органов и дыхательных путей у детей // Практика педиатра. 2023. № 1. С. 47-50.
Комментарии
ПРАКТИКА ПЕДИАТРА



