Роль Глюкофажа в патогенетическом лечении сахарного диабета 2 типа
СтатьиОпубликовано в журнале:
РМЖ Том 18, № 10, 2010
К.м.н. И.В. Кононенко, профессор О.М. Смирнова
ФГУ Эндокринологический научный центр, Москва
Сахарный диабет 2 типа – хроническое заболевание обмена веществ, характеризующееся стойкой гипергликемией, которая является результатом дефектов секреции и действия инсулина. Это тяжелое, хроническое и постоянно прогрессирующее заболевание. Неблагоприятный прогноз у больных сахарным диабетом 2 типа (СД 2 типа) определяется развитием макро– и микрососудистых осложнений. Причиной макрососудистых осложнений является атеросклеротическое поражение основных артериальных бассейнов, приводящее к развитию ишемической болезни сердца и ее осложнений, цереброваскулярной болезни и облитерирующего поражения артерий нижних конечностей. В основе микрососудистых осложнений лежит специфическое для СД поражение микроциркуляторного русла, связанное с утолщением базальных мембран капилляров. Клинические проявления микроангиопатии – диабетическая нефропатия и ретинопатия. СД – самая частая причина слепоты у взрослых. Целью лечения сахарного диабета является нормализация уровня гликемии и снижение риска развития макро– и микрососудистых осложнений. Важнейшие факторы риска, оказывающие влияние на развитие сердеч но–сосудистых осложнений у больных СД 2 типа – состояние углеводного обмена, уровень артериального давления и липидный спектр плазмы крови. В таблице 1 представлены целевые значения основных показателей, достижение которых обеспечивает эффективность лечения больных СД 2 типа.
Таблица 1. Контрольные параметры (цели лечения) при сахарном диабете 2 типа (Алгоритмы специализированной помощи больным сахарным диабетом, 2009 г.)
| HbA1c | <7,0% |
| Глюкоза натощак | <6,5 ммоль/л |
| Глюкоза через 2 часа после еды | <8,0 ммоль/л |
| Артериальное давление | 130/80 мм рт.ст. |
| Общий холестерин | <4,5 ммоль/л |
| ХС ЛПНП | <2,6 ммоль/л |
| ХС ЛПВП у мужчин | >1,0 ммоль/л |
| ХС ЛПВН у женщин | >1,2 ммоль/л |
| Триглицериды | <1,7 ммоль/л |
В середине 60–х годов XX века возникла основная концепция механизма развития СД 2 типа [1,2]: ожирение (в особенности висцеральный тип ожирения) сопровождается инсулинорезистентностью → длительно существующая инсулинорезистентность вызывает компенсаторную гиперинсулинемию → гиперфункция β–клеток приводит со временем к нарушению секреции инсулина, к развитию относительного, а затем абсолютного дефицита инсулина и развитию гипергликемии. Инсулинорезистентность (ИР) и дисфункция β–клеток – главные эндокринные дефекты в развитии СД 2 типа. Само по себе наличие инсулинорезистентности не способно привести к развитию СД 2 типа. Необходимым условием для развития и прогрессирования СД 2 типа является наличие выраженной дисфункции β–клеток поджелудочной железы.
Очевидно, что оба эти процесса нельзя рассматривать в отрыве один от другого. ИР присутствует у большинства больных с ожирением и может ускорять прогрессирование диабета у людей с дисфункцией β–клеток. Результаты проведенных исследований по изучению возможности профилактики СД 2 типа позволили сделать вывод, что нефармакологические мероприятия и фармакологические средства в основном влияют на ИР. Снижение уровня ИР приводит к поддержанию и защите функции β–клеток [3].
Под инсулинорезистентностью понимают нарушение биологического действия инсулина при его достаточной концентрации в крови. Эти изменения касаются как метаболических процессов (обмена углеводов, жиров и белков), так и митотических процессов (нарушение роста, дифференцировки клеток, синтеза ДНК, регуляции транскрипции генов). Механизмы различных биологических эффектов инсулина разветвляются на биохимических стадиях, следующих после связывания инсулина с рецептором. Тирозинкиназа инсулинового рецептора фосфорилирует много субстратов и каждый фосфорилированный белок запускает разные биологические процессы [4].
При СД 2 типа наблюдается резистентность мышечной, жировой ткани, а также ткани печени к действию инсулина.
ИР мышечной ткани определяется генетической предрасположенностью и может быть выявлена задолго до клинической манифестации СД 2 типа. Нарушение инсулинообусловленного усвоения глюкозы в мышечной ткани связано с дефектами транспорта и фосфорилирования глюкозы и, как следствие, с нарушением синтеза гликогена [5].
10–20% глюкозы усваивается жировой тканью. Блокада глюкозных транспортеров GLUT4, специфичных для жировой ткани, вызывает ИР всего организма, в то время как блокада глюкозных транспортеров GLUT4 в скелетных мышцах приводит к усилению усвоения глюкозы жировой тканью [6]. ИР и ожирение в большинстве случаев сопровождают друг друга. Что изначально генетически предопределено – ожирение или снижение чувствительности тканей к инсулину – остается предметом дискуссий.
Жировая ткань состоит из адипоцитов, преадипоцитов, нервов, макрофагов, фибробластов, сосудов. Адипоцит может максимально запасать 0,8 μg липидов в клетке. Увели чение размера адипоцита сопровождается инсулинорезистентностью, снижением внутри него метаболизма глюкозы и триглицеридов. Вследствие отсутствия адекватного ангиогенеза при гипертрофии адипоцитов отмечается уменьшение перфузии жировой ткани. Отсутствие адекватного кровотока в гипертрофированной жировой ткани приводит к гипоксии последней, что тормозит процессы дифференцировки адипоцитов, экспрессии адипонектина, способствует формированию свободных радикалов, воспалению и гибели клеток. При этом вокруг адипоцитов усиливается миграция макрофагов [7]. Оксидативный стресс в жировых клетках вследствие снижения перфузии и относительной гипоксии приводит к состоянию хронического воспаления. Локально отмечается увеличение концентрации медиаторов воспаления и цитокинов, накопление триглицеридов. Эпикардиальный, внутримышечный и висцеральный абдоминальный жир метаболически более активен и менее чувствителен к влиянию инсулина. В висцеральной жировой ткани отмечено усиление синтеза фактора некроза опухоли–альфа (TNF–α), который снижает активность тирозинкиназы инсулинового рецептора и фосфорилирование белков – субстратов инсулинового рецептора. Неспособность инсулина, вследствие инсулинорезистентности жировой ткани, подавлять окисление липидов приводит к высвобождению большого количества свободных жирных кислот (СЖК). Повышение уровня СЖК вызывает ингибирование процессов транспорта и фосфорилирования глюкозы, снижает окисление глюкозы и синтез гликогена в мышцах [8]. Перемещаясь из адипоцитов в склетные мышцы, СЖК участвуют в формировании свободных радикалов во время окислительного фосфорилирования, приводят к внутриклеточному накоплению токсических метаболитов жирового обмена (диацилглицерола, ацетил–коэнзима А, керамидов), что вызывает оксидативное повреждение и нарушает передачу сигнала через инсулиновый рецептор. Высказано предположение, что окисление липидов может наблюдаться генерализовано и регионарно, например, в миокардиоцитах, в скелетных мышцах, в печени. Избыток СЖК в печени приводит к синтезу атерогенных липопротеинов (ЛПНП и ЛПОНП). Последние исследования показали способность СЖК вызывать дисфункцию β–клеток и окислительный стресс в них.
В печени СЖК препятствуют разрушению инсулина, в результате чего периферические ткани, мозг, сосуды подвергаются гиперинсулинемии, что усугубляет инсулинорезистентность и вызывает активацию симпатической нервной системы [9]. В головном мозге СЖК вовлечены в механизмы центральной регуляции синтезы глюкозы.
В настоящее время жировая ткань рассматривается как эндокринный орган, обеспечивающий поступление в кровоток большого количества гормонально активных веществ (лептина, адипонектина, резистина, апелина, эндоканнабиноидов), свободных жирных кислот и цитокиновов (TNF–α, IL–6).
Продукция жировой тканью канабиноидов уменьшает экспрессию адипонектина, снижает выделение энергии в жировой ткани и окисление жиров, усиливает синтез последних. Было показано, что инсулин ингибирует синтез эндоканабиоидов и резистентность жировой ткани к инсулину может быть причиной увеличения уровня эндоканабиноидов, что, в свою очередь, влияет на регуляцию углеводного обмена, аппетит, энергетический обмен.
Возможно, что ИР жировой ткани является ранним и необратимым дефектом и может объяснять причинную взаимосвязь между нарушением функции адипоцита и ИР мышечной ткани и гепатоцитов. Происхождения этого взаимодействия имеет генетическую основу и закладывается в период эмбриогенеза, задолго до развития метаболических нарушений [10]. Внутриутробное питание и гормональные факторы, низкий вес новорожденного определяют риск развития метаболических и сердечно–сосудистых заболеваний у взрослого. Исследования показали, что снижение веса приводит к нормализации ИР скелетных мышц, печени, АД, дислипидемиии и гипергликемии, но не изменяет ИР жировой ткани и гипоперфузию в ней. Нарушение транслокации глюкозного транспортера GLUT4 в миоцитах является обратимым процессом, но не изменяется в жировой ткани. Резистентность жировой ткани к катехоламинам также остается неизменной при снижении веса.
Инсулин способен проникать через гематоэнцефалический барьер [11]. Рецепторы к инсулину обнаружены в центральной нервной системе (ЦНС) в гипоталамусе. Инсулин оказывает влияние на аппетит и потребление пищи, напрямую подавляя транскрипцию mRNA нейропептида Y, усиливая экспрессию проопиомеланокортина в аркуатных ядрах гипоталамуса. Также было обнаружено, что введение инсулина в область третьего желудочка приводит к подавлению продукции глюкозы печенью. По–видимому, механизм действия инсулина опосредован через центральные АТФ–зависимые К+–каналы и активацию автономной нервной системы. О значимой роли рецепторов к инсулину в центральной нервной системе свидетельствует тот факт, что у мышей с нарушением передачи инсулинового сигнала в ЦНС наблюдалась более выраженная гипергликемия, чем у мышей, имеющих нарушения инсулинового сигнала на периферии. В связи с этим высказывается предположение, что, как и нарушения метаболизма глюкозы, так и ожирение могут быть следствием нарушений передачи инсулинового сигнала в гипоталамусе.
Печень играет главную роль в регуляции гомеостаза глюкозы, в обмене жирных кислот и аминокислот [12]. Это основой источник эндогенной продукции глюкозы, основное место, где происходит этерификация и окисление жирных кислот. Здесь же происходит разрушение инсулина. ИР ткани печени приводит к увеличению продукции глюкозы печенью преимущественно – за счет глюконеогенеза, в меньшей степени за счет распада гликогена. Процессы гликогенолиза и глюконеогенеза напрямую зависят от концентрации инсулина в крови и эффективности его действия. При возникновении относительной или абсолютной недостаточности инсулина, а также при развитии ИР они становятся активны, что приводит к резкому увеличению выработки глюкозы печенью и формированию гипергликемии. В эксперименте у мышей, лишенных рецепторов к инсулину в печени наблюдалась стойкая гипергликемия натощак и после еды, периферическая ИР. Отсут ствие же рецепторов к инсулину в жировой и мышечной ткани не приводила в каждом случае к развитию гипергликемии.
Таким образом, ИР жировой, мышечной ткани и клеток печени является основной генетически обусловленной составляющей патогенеза СД 2 типа и тесно связана с другими факторами риска сердечно–сосудистых заболеваний, такими как ожирение, дислипидемия, артериальная гипертензия.
Вот уже более полувека в клинической практике для лечения сахарного диабета используется препарат из класса бигуанидов – метформин. Основной механизм действия метформина направлен на уменьшение инсулинорезистентности печеночной ткани и снижение избыточной продукции глюкозы печенью. Метформин подавляет глюконеогенез, блокируя ферменты данного процесса в печени, снижает гликогенолиз. Прием 1000–2550 мг метформина в сутки в течение нескольких месяцев [13,14] снижал базальный уровень продукции глюкозы печенью на 9–30% по сравнению с исходными данными и плацебо, что тесно коррелировало со снижением уровня глюкозы плазмы натощак. В присутствии инсулина метформин также увеличивает периферическую утилизацию глюкозы мышцами, активируя тирозинкиназу инсулинового рецептора и транслокацию ГЛЮТ4 и ГЛЮТ1 в мышечной и жировой тканях. Метфор мин улучшает анаэробное окисление глюкозы, что сопровождается повышением постпрандиального уровня молочной кислоты, усиливает синтез гликогена в скелетных мышцах. Метформин повышает утилизацию глюкозы кишечником (усиливая анаэробный гликолиз), что проявляется в снижении уровня глюкозы в крови, оттекающей от кишечника.
Длительное применение метформина положительно влияет на липидный обмен: приводит к снижению уровня триглицеридов, общего холестерина, холестерина липопротеидов низкой плотности в сыворотке крови [15].
Механизм действия метформина – антигипергликемический, а не гипогликемический. Метформин не снижает содержание глюкозы в крови ниже ее нормального уровня – вот почему при монотерапии метформином отсутствуют гипогликемические состояния. Препарат не изменяет уровня секреции инсулина и не оказывает сахароснижающего эффекта при абсолютном дефиците инсулина.
Метформин обладает умеренно выраженным анорексигенным эффектом, что, возможно, связано с центральным действием препарата на уровне гипоталамических нейронов [16]. У больных, получающих метформин, наблюдается стабилизация или снижение массы тела, преимущественно за счет уменьшения висцеральной жировой ткани [17]. Препарат уменьшает гиперинсулинемию у больных, страдающих ожирением. Доказано положительное влияние метформина и на фибринолитические свойства крови за счет подавления ингибитора активатора плазминогена–1.
Метформин является препаратом, прием которого достоверно снижает общую частоту макро– и микрососудистых осложнений СД 2 типа, что отражается на продолжительности жизни больных. Результаты проспективного исследования, проведенного в Великобритании UKPDS (UK Prospective Diabetes Study), показали, что применение метформина (Глюкофажа) с момента установления диагноза снижает на 42% смертность от причин, связанных с сахарным диабетом, общую смертность на 36%, а частоту диабетических осложнений на 32% [18,19]. С 2005 г. в рекомендациях Международной диабетической федерации (International Diabetes Fede ration – IDF) метформин является препаратом первого выбора для лечения сахарного диабета 2 типа [20]. С 2006 года в рамках согласованного алгоритма Американской (ADA) и Европейской диабетологической ассоциаций (EASD) назначение метформина показано сразу же при постановке диагноза СД 2 типа одновременно с изменением образа жизни (рис. 1) [21].
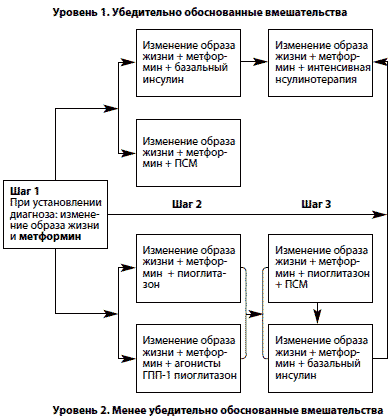
Рис. 1. Согласованный алгоритм лечения сахарного диабета 2 типа (ADA/EASD 2006 г.)
В UKPDS исследовании при изучении эффективности метформина в лечении СД 2 типа использовался препарат Глюкофаж. Средняя суточная доза препарата для большинства пациентов составляла 2000 мг. Именно при такой дозировке препарата наблюдалось максимальное снижение уровня HbA1c (на 2%), причем количество побочных эффектов со стороны желудочно–кишечного тракта было не выше, чем при дозе 500 мг/сут. [22].
Механизмы обнаруженного в исследованиях UKPDS благоприятного влияния Глюкофажа на сердечно–сосудистые показатели до сих пор изучаются. Исследования in vitro показали, что Глюкофаж нарушает адгезию моноцитов, их трансформацию в макрофаги и способность захватывать липиды. Такая активность в условиях in vivo может оказывать антиатерогенное действие. Глюкофаж ослабляет гликозилирование белков. Диаминовый компонент Глюкофажа связывает и нейтрализует активные вещества, образующиеся на промежуточных этапах процессов гликозилирования белков. Изучается влияние препарата на микроциркуляцию, в том числе на увеличение количества капилляров в ишемизированной ткани и усиление кровотока (табл. 2) [23].
Таблица 2. Влияние Глюкофажа на риск развития сердечно–сосудистых осложнений сахарного диабета
| Улучшает | Ослабляет |
|---|---|
| Чувствительность к инсулину Липидный профиль Фибринолиз Микроциркуляцию Кровоток в ишемизированной ткани Реологию крови |
Гипертриглицеридемию Перекрестное сшивание фибрина Неоваскуляризацию Окислительный стресс Экспрессию молекул адгезии на эндотелиоцитах Процессы дифференцировки клеток воспаления в макрофаги |
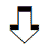 |
|
| Снижает риск развития сердечно–сосудистых осложнений сахарного диабета | |
В ретроспективном популяционном исследовании применение метформина (изолированно или в сочетании с препаратами сульфонилмочевины) сопровождалось снижением риска смерти по сравнению с применением препаратов сульфонилмочевины [24]. В течение среднего срока наблюдения 5,1 лет смертность среди больных, получавших препараты сульфонилмочевины, метформин или их комбинацию и не получавших инсулин, составила 24,7, 13,8 и 13,6% соответственно.
Результаты UKPDS исследования показали, что Глюкофаж обладает антигипергликемическим действием, аналогичным по эффективности действию производных сульфонилмочевины, не вызывая при этом увеличения массы тела. Монотерапия Глюкофажем позволила снизить уровень HbA1c в среднем на 1,4% в течении 29 недель [25].
При сравнении эффективности пиоглитазона и метформина у 205 больных с недавно диагностированным СД 2 типа было показано, что оба препарата при монотерапии в течение 32 недель одинаково эффективно снижали уровень глюкозы (по уровню глюкозы натощак и HbA1c) [26]. Действие пиоглитазона осуществлялось в основном за счет усиления чувствительности периферических тканей к инсулину, в то время как метформина – за счет снижения продукции глюкозы печенью. На фоне терапии пиоглитазоном отмечалось более значимое увеличение общей чувствительности тканей к инсулину. Применение пиоглитазона сопровождалось небольшим увеличением массы тела, а применение метформина – небольшим снижением. При применении метформина чаще наблюдалась диарея, а при применении пиоглитазона – отек нижних конечностей.
Побочные действия со стороны желудочно–кишечного тракта нередко являются основным препятствием в назначении Глюкофажа. В рандомизированных исследованиях около 30–40% больных при назначении метформина отмечают те или иные нежелательные явления со стороны ЖКТ: диарею, метеоризм, боли в животе [27,28]. Однако несмотря на это, лечение метформином было прервано только в 5% случаев [29]. Нежела тельных эффекты со стороны ЖКТ удается избежать путем снижения начальной дозы препарата (500 мг/сут. в течение недели), медленными темпами дальнейшей ее титрации. Недавно в целях уменьшения нежелательных эффектов со стороны ЖКТ была разработана новая таблетированная форма метформина – метформин с пролонгированным высвобождением (Глюкофаж Лонг®). Применение новой формы препарата позволит улучшить переносимость метформина [30].
При отсутствии адекватного гликемического контроля на фоне терапии пероральными сахароснижающими препаратами пациентам с СД 2 типа показана инсулинотерапия. Однако даже на фоне инсулинотерпии у больных с избыточной массой тела или ожирением достижение компенсации СД остается трудной задачей. Одной из причин этого является сохраняющаяся инсулинорезистентность и прибавка массы тела на фоне инсулинотерапии. В подобных случаях комбинированная терапия Глюкофажем и инсулином может значительно улучшить компенсацию углеводного обмена. Резуль таты исследования показали, что при достижении практически одинакового уровня HbA1c (7,5% в группе инсулин + метформина и 7,9% в группе инсулин + плацебо) у больных, получавших плацебо, отмечались более высокие дозы инсулина (84 ед. и 67 ед. в сутки соответственно) и прибавка массы тела, в то время как на фоне приема метформина прибавки массы тела не установлено [31].
Проведенное пилотное исследование показало, что метформин не оказывает отрицательного влияния на состояние печени при неалкогольном стеатогепатите [32]. Больные с неалкогольным стеатогепатитом, не страдающие СД 2 типа, но имеющие периферическую инсулинорезистентность, были разделены на 2 группы путем рандомизации. Всем больным в качестве лечебного питания была рекомендована диета, богатая овощами, фруктами, с низким содержанием насыщенных жирных кислот и холестерина, снижение веса и дозированная физическая нагрузка. Больным первой группы был назначен метформин 1000 мг/сут. в течение 12 мес. Результаты гистологического анализа ткани печени не выявили различий в группах больных неалкогольным стеатогепатитом получавших метформин и без него. При этом в обеих группах отмечалось достоверное значимое снижение уровня печеночных трансаминаз (ALT, AST) и улучшение состояния клеток печени.
Строгие критерии ограничения применения метформина при СД 2 типа основываются на полученных в 1970–х годах сведениях о смертности от лактат–ацидоза, связанного с применением фенформина. Соответ ствующие данные для метформина крайне редки [33]. Как известно, действие метформина связано с усилением усвоения глюкозы периферическими тканями. При этом он снижает активность ферментов пируватдегидрогеназного комплекса, направляя тем самым окисление глюкозы по анаэробному пути до образования молочной кислоты. При нормальной функции почек из быток молочной кислоты легко удаляется с мочой. При снижении фильтрационной способности почек и метформин, и молочная кислота могут задерживаться в организме. Развитие молочнокислого ацидоза представляет серьезную угрозу для жизни. Однако увеличивает ли именно метформин риск молочнокислого ацидоза, является спорным вопросом. Кохрановский анализ 200 исследований показал, что применение метформина не приводит к увеличению случаев лак тат–ацидоза [34]. Было установлено, что в группе больных СД 2 типа, получавших метформин, случаи лактат–ацидоза составили 5,1 на 100 тыс. больных, в то время как в группе не получающих метформин – 5,8 на 100 тыс. Bodmer с соавт. [35], проанализировав терапию 50 048 больных СД 2 типа, зафиксировали, что в группе получающих метформин случаи лактат–ацидоза составили 3,3 на 100 тыс. больных, в группе же получавших препараты сульфонилмочевина – 4,8 на 100 000 больных. При этом во всех случаях развития лактат–ацидоза у больных наблюдались состояния, сопровождающиеся высоким риском развития лактат–ацидоза. Факторами риска развития лактат–ацидоза являются состояния, которые способствуют либо усилению продукции молочной кислоты, либо снижают ее выведение: застойная сердечная недостаточнось, заболевания печени, шок, употребление алкоголя, со стояния, сопровождающиеся гипоксией (дыхательная и сердечная недостаточность, анемия), почечная недостаточность, сепсис. Учитывая, что Глюкофаж выделяется почками, при назначении препарата и в дальнейшем в ходе лечения необходимо оценивать фильтрационную способность почек. Противопока занием для использования метформина является уровень креатинина более 132 мкмоль/л у мужчин и 123 мкмоль/л у женщин или клиренс креатинина <60 мл/мин. Особого внимания требуют пожилые пациенты, которые часто имеют пониженную функцию почек и сопутствующие заболевания сердечно–соудистой системы, органов дыхания, анемию. Пожилым больным не рекомендуется назначать максимальные терапевтические дозы препарата.
Глюкофаж – единственный из антидиабетических препаратов, который прошел клинические испытания и утвержден к применению у детей старше 10 лет, страдающих СД 2 типа. Разрешенная доза препарата составляет 1000–2000 мг/сут.
В ряде рандомизированных контролируемых исследований был продемонстрирован дозозависимый эффект метформина. Увеличение суточной дозировки препарата приводило к усилению сахароснижающего эффекта [36]. Среднеэффективной дозой считается 2000 мг/сут., максимально допустимой – 3000 мг/сут.
Доказанная эффективность в лечении и профилактике сахарного диабета, уменьшение неблагоприятных кардиоваскулярных исходов на фоне терапии, относительная безопасность метформина даже при использовании высоких доз, невысокая стоимость препарата – все это делает метформин препаратом первого выбора в лечении СД 2 типа.
- Reaven G.M. Banting lecture 1988/ Role of insulin resistance in human disease. Diabetes.1988.–V/37.P.1595–1607.
- DeFronzo R.A. Lilly Lecture. The triumvirate: beta cell, muscle, liver. A collusion responsible for NIDDM. // Diabetes. – 1988. – Vol. 37. – P. 667–687.
- Preservation of pancreatic beta–cell function and prevention of type 2 diabetes by pharmacological treatment of insulin resistance in high–risk Hispanic women. Diabetes 51: 2796–2803, 2002.
- Молекулярная эндокринология. Под редакцией Брюса Д.Вайнтрауба–М.: Медицина.–2003. –стр.277–291.
- Tuomilheto J., Lindstrom J., Eriksson J.G., et al. 2001.
- Cahova M., Vavrinkova H.Glucose–fatty acid interaction in skeletall muscle and adipose tissue in insulin resistance. Physiol Res 2007; 56:1–15 7. Lumeng CN, Deyoung SM, Bodzin JL. Increased inflammatory properties of adipose tissue macrophages recruited during diet–induced obesity. Diabetes 2007; 56:16–23 8. Hennes M.M., Shrago E., Kissebah A ,1998.
- Patrica Iozzo. Viewpoints on the way to the consensus session. Where does insulin resistanc start? The adipose tissue. Diabetes Care 2009; V.32. Suppl.2 S168–173.
- Nelson T.L., Vogler G.P., Pedersen N.L. Genetic and environment influences on body fat distribution? Fasting insulin levels and CVD: are the influences shared? Twin Res 2003; 3;43–50/ 11. Uberto Pagotto. Where does insulin resistanc start? The brain. Diabetes Care 2009; V.32. Suppl.2 S174–177.
- Gianluca Perseghin. Viewpoints on the way to the consensus session. Where does insulin resistanc start? The liver. Diabetes Care 2009; V.32. Suppl.2 S164–167.
- Jackson R.A., Hawa M.L., Jaspan J.B. et al 1987; Mechanism of metformin action in non–insulin– dependent diabetes. Diabetes 1997; 36:632–640.
- Cusi K., Consoli A., DeFronzo R.A. Metabolic effects of metformin on glucose and lactate metabolism in noninsulin–dependent diabetes mellitus. J Clin Endocrinol Metab 1996; 81:4059–4067.
- Jappesen J., Zhou MY., Chen YD., Reaven G.M. Tffect of metformin on postprandial lipemia in patients with fairly to poorly controlled NIDDM. Diabetes Care 1994;17:1093–1099.
- Chau–Van C., Gamba M., Salvi R. et al. 2007.
- Charles M.A., Morange P., Eschwege E. 1998.
- UK Prospective Diabetes Study (UKPDS) Group. UK prospective diabetes study 16. Overview of 6 years’ therapy of type II diabetes: a progressive disease. // Diabetes. – 1995. – V.44. – P. 1249–1258.
- UK Prospective Diabetes Study (UKPDS) Group. // The Lancet . – 1998. – V.352. – P.837–853.
- International Diabetes Federation, 2005. Global guideline for type 2 diabetes.
- Nathan DM, Buse JB,Davidson MB et al. American Diabetes Association, European Association for the Study of Diabetes. Medical management of hyperglycemia in type 2 diabeyes: a consensus algorithm for the initiation and adjustment of therapy: a consensus statement of the American Diabetes Association and European Association for the Study of Diabetes. Diabetes Care. 2009;32:193–203.
- Garber et al. Am J Med. 1997; 103:491–497.
- Cardiovascular benefits of metformin. Diabetes and metabolism. 2003 september.
- Jonson J.A. et al. Diabetes Care, 2002; 25:2244– 2248.
- Hermann L.S., Schersten B., Bitzen P.O. et al. Therapeutic comparison of metformin and sulfonylurea along and in various combinations. A double–bind controlled study. Diabetes Care 1994; 17:1100–1109.
- J Clin Endocrinol Metab, 2003; 88: 1637–1645.
- Kahn SE, Haffner SM et al. Glycemic durability of rosiglitasone, metformin or glyburide monoterapy. N Engl J Med 2006; 355: 2427–43.
- Davidson JA, Scheen AJ, Howlett HC. Tolerability profile of metformin/glibenclamid combinationtabletes (Glucovance). Drug Saf 2004;27: 1205–16.
- Ramsdell LW, Grossman JA, Stephens JM. A shot–term cost–of–treatment model for type 2 diabetes: comparison of glipizide gastrointestinal therapeutic system, metformin, and acarbose. Am J Maneg Care 1999; 5:1007–1024.
- Michael D Feher; Ma’en Al–Mrayat. Tolerability of Prolonged–Release Metformin (Glucophage) in individuals intolerant to standard metformin–results from four UK centres. British J Diabetes and Vascular Diseade. 2007; 7 (5): 225–228.
- Kooy A, de Jager J, Lehert P, et al. Arch Intern Med. 2009; 169:616–625.
- William W.Shields; K.E.Thompson; G.A. Grice et al. The effect of metformin and standard therapy versus standard therapy alone in nondiabetic patients with insulin resistence and nonalcoholic steatohepatitis (NASH): a pilot trial. Ther Adv Gasyroenterol.2009; 2(3): 157–163.
- Rachmani R., Slavachevski I., Levi Z., Zadok B. 2002.
- Salpeter SR., Greyber E., Pasternak GA., Salpeter EE. 2 Risk of fatal and nonfatal acidosis with metformin use in type 2 diabetes mellitus. Cochrane Database syst Rev. 2006; 1 CD002967.
- Bodmer M., Meier C., Krahenbuhl S. et. al.Metformin, sulfonilureas, or other antidiabetes drugs and the risk of lactic acidosis or hypoglycemia a nested case–control analysis. Diabetes Care. 2008;31: 2086–2091.
- Garber AJ, Duncan TG, Goodman AM. Et al. Efficacy of metformin in type 2 diabetes results of a double–blind, placebo–controlled, dose–response trial. Am J Med 1997; 103: 491–497.



