Исключительная коморбидность двух патологий (инфантильная форма болезни Помпе и врожденный порок сердца)
СтатьиН.П. Котлукова1 2, д-р мед. наук, профессор, Е.В. Внукова1, О.А. Окулова2, Н.Д. Тележникова2
1 ФГАОУ ВО «Российский национальный исследовательский медицинский университет им. Н.И. Пирогова» Минздрава России, г. Москва
2 ГБУЗ «Детская городская клиническая больница им. З.А. Башляевой Департамента здравоохранения г. Москвы»
Ключевые слова: болезнь Помпе, врожденный порок сердца, болезнь накопления, ген GAA, заместительная ферментная терапия
Keywords: Pompe disease, congenital heart defect, storage disease, GAA gene, enzyme replacement therapy
Резюме. Болезнь Помпе, также известная как гликогеноз типа II или гликогенная миопатия, представляет собой орфанное заболевание с накоплением гликогена в клетках из-за дефицита фермента а-1,4-глюкозидазы. Клиническая картина болезни варьирует в зависимости от возраста пациента и тяжести течения, что затрудняет диагностику. Она может проявляться в тяжелой инфантильной форме, характеризующейся поражением сердца и скелетных мышц у детей, или более легкой, проявляющейся у взрослых миопатическим синдромом. Для своевременной постановки диагноза болезни Помпе используются различные методы, включая стандартные лабораторные и инструментальные исследования, энзимодиагностику и генетическое тестирование. Ключевым моментом ведения пациента является раннее начало заместительной ферментной терапии рекомбинантной а-1,4-глюкозидазой (rhGAA), а также новым препаратом следующего поколения - авалглюкозидазой-альфа.
В данной статье представлен клинический случай пациента с сочетанным течением младенческой формы болезни Помпе и врожденного порока сердца, который был успешно диагностирован на ранних стадиях и получил эффективное лечение. Этот случай подчеркивает важность ранней диагностики, своевременного начала терапии и междисциплинарного подхода в лечении пациентов с подобными состояниями.
Summary. Pompe disease, also known as glycogen storage disease type II or glycogenic myopathy, is an orphan disorder characterized by glycogen accumulation in cells due to deficiency of the enzyme acid alpha glucosidase. The clinical presentation of the disease varies with patient age and disease severity, which complicates diagnosis. It can present as a severe infantile form, characterized by cardiac and skeletal muscle involvement in children, or as a milder adult-onset myopathic syndrome. Various methods are used for timely diagnosis of Pompe disease, including routine laboratory and instrumental studies, enzyme testing, and genetic analysis. A key aspect of patient management is early initiation of enzyme replacement therapy with recombinant human acid alpha glucosidase, as well as treatment with the next generation agent avalglucosidase alfa. This article presents a clinical case of a patient with coexisting infantile Pompe disease and a congenital heart defect who was successfully diagnosed at an early stage and received effective treatment. This case underscores the importance of early diagnosis, timely initiation of therapy, and an interdisciplinary approach in the care of patients with such conditions.
Для цитирования: Исключительная коморбидность двух патологий (инфантильная форма болезни Помпе и врожденный порок сердца) / Н.П. Котлукова, Е.В.Внукова, О.А.Окулова, Н.Д.Тележникова // Практика педиатра. 2025. № 3. С. 12-19.
For citation: Exceptional comorbidity of two pathologies (infantile form of Pompe disease and congenital heart disease) / N.P. Kotlukova, E.V. Vnukova, O.A. Okulova, N.D. Telezhnikova // Pediatrician's Practice. 2025;(3): 12-19. (In Russ.)
ВВЕДЕНИЕ
Болезнь Помпе (БП) (гликогеноз II типа) - редкое наследственное заболевание, вызванное дефицитом лизосомальной а-1,4-глюкозидазы [1]. При нем происходит накопление гликогена в различных тканях организма, особенно в мышцах и сердце. Клинически заболевание проявляется генерализованной быстропрогрессирующей мышечной гипотонией, дыхательной недостаточностью (ДН), гипертрофической кардиомиопатией с развитием жизнеугрожающей сердечной недостаточности (СН), гепатомегалией [2]. Выделяют две основные формы: инфантильную с манифестацией в первые месяцы жизни, летальным исходом на первом году жизни при отсутствии терапии и БП с поздним началом, которая развивается в любом возрасте старше одного года, имеет более легкое течение и более благоприятный прогноз [3].
Болезнь Помпе была впервые описана в 1932 г. голландским врачом Йоханом Каспером Помпе. Его открытие произошло в результате работ по изучению случая 7-месячной девочки с необычными симптомами, включая мышечную слабость и увеличение печени и сердца. После проведения аутопсии у погибшего ребенка Помпе и его коллеги обнаружили накопление гликогена в различных тканях, что и привело к открытию нового заболевания [4]. Ген GAA, отвечающий за синтез фермента, был клонирован и секвенирован в 1991 г., а в 2006 г. одобрена первая заместительная ферментная терапия для лечения пациентов с этим заболеванием [1].
Широкий спектр клинических проявлений БП, высокая вариабельность возраста манифестации затрудняют диагностику, при этом раннее начало терапии напрямую связано с ее эффективностью. Изучение болезни Помпе и ее сочетание с другими патологиями демонстрируют важность многопрофильного подхода в диагностике и лечении. Представленный нами случай является исключительным, поскольку в литературе нет описанных случаев ведения пациентов с БП и врожденным пороком сердца. За десятилетия исследований и деятельности медицинских специалистов накоплен богатый опыт ведения пациентов с этим заболеванием, что приводит к улучшению качества их жизни и разработке новых методов терапии.
Эпидемиология
На данный момент сообщается, что частота встречаемости заболевания намного больше общепринятых значений 1 : 40 000. По данным неонатального скрининга 11,6 млн новорожденных с четырех континентов, частота встречаемости БП составляет 1 : 18 711 рожденных вне зависимости от географического региона и этнической принадлежности. Младенческая (ювенильная) форма БП встречается 1 : 126 118 рожденных, в то время как взрослая форма заболевания - 1 : 21 902 рожденных [4]. Частота БП в России остается неизвестной.
Генетика
Болезнь Помпе (OMIM № 232300) - редкое аутосомнорецессивное заболевание, которое относится к группе болезней накопления. Это состояние обусловлено мутацией в гене GAA, который расположен на хромосоме 17q25.2-25.3. Он состоит из 20 экзонов и имеет размер 20 Кб [1]. Ген GAA кодирует кислую а-1,4-глюкозидазу из группы лизосомальных гидролаз. Синтез фермента происходит в эндоплазматическом ретикулуме, затем с помощью рецептора манно-зо-6-фосфата направляется в лизосому, где обеспечивает деградацию гликогена [1]. Мутация в данном гене приводит к дефициту или нулевому количеству фермента, в связи с чем гликоген накапливается в лизосомах, что запускает метаболический каскад с разрушением тканей и органов организма [3]. Наиболее подвержены этому процессу мышечные ткани (сердечная, скелетная и гладкая мускулатура).
На данный момент в базе данных вариантов GAA болезни Помпе описано 911 мутаций гена, из них 648 патогенных, приводящих к нарушению синтеза фермента, а 263 считаются генетическими вариантами неизвестного значения [5]. Как и для большинства генетических заболеваний, сложно установить гено-фенотипическую корреляцию. Однако можно сделать общие выводы. Уровень активности а-1,4-глюкозидазы положительно коррелирует с возрастом манифестации и скоростью прогрессирования заболевания [1].
Миссенс-варианты и варианты сплайсинга могут приводить к полному или частичному (2-40% от уровня нормы) отсутствию активности фермента [1]. При этом наблюдается широкая клиническая вариабельность заболевания. Так, в одной семье родственники с одинаковыми патогенными мутациями могут наблюдаться как с младенческой, так и с взрослой формой заболевания. Вероятно, существуют модифицирующие факторы, которые влияют на течение заболевания, но эта гипотеза требует дальнейших исследований.
При наличии нонсенс-мутаций которые приводят к практически нулевой активности фермента (<1% от уровня нормы) у пациента чаще всего развивается инфантильная форма с тяжелым течением [1].
Некоторые варианты мутаций встречаются чаще, чем другие в отдельных популяциях. Примеры приведены в таблице 1.
Таблица 1. Корреляция генотипа и фенотипа в отдельных популяциях [1, 4, 5]
|
Генотип |
Частота встречаемости |
Фенотип |
|
c.2560C>T (p.Arg854Ter) |
50-60% африканцев или афроамериканцев |
Инфантильная форма БП с тяжелым течением |
|
c.525del (p.(Glu176Argfs*45)) |
34% в Нидерландах |
Инфантильная форма БП с тяжелым течением |
|
c.2481 + 102_2646 + 31del (p.(Gly828_ Asn882del)) (делеция 18-го экзона) |
25% в Нидерландах |
|
|
c.1935C>A (p.Asp645Glu) |
40-80% в Тайване и Китае |
Младенческая формы БП |
|
c.-32-13T>G (IVS1) |
50-85% больных в европеоидной расе |
Не связан с конкретной формой заболевания |
Кроме того, известно 148 вариантов мутаций «псевдодефицита», они снижают уровень синтеза фермента, но не вызывают заболевание, что приводит к ложноположительному диагнозу [1, 5].
Клиническая картина и диагностика
Выделяют две основные формы заболевания: инфантильную (классическую, раннюю, младенческую) и позднюю (юношескую, взрослую), в зависимости от возраста манифестациии, тяжести течения и скорости прогрессирования [3] (табл. 2). Обычно эти показатели имеют положительную корреляцию между собой, что важно для определения прогноза.
Таблица 2. Клиническая картина [3, 6]
|
Форма заболевания |
Инфантильная форма |
Взрослая форма |
|
Уровень активности фермента |
<1% |
2-40% |
|
Возраст манифестации |
0-12 мес |
Любой возраст старше 12 мес |
|
Астеновегетативный синдром |
• Слабость |
|
|
Опорно-двигательный аппарат |
• Прогрессирующая мышечная слабость и гипотония |
• Прогрессирующая мышечная слабость и гипотония |
|
Дыхательная система |
• Дыхательная недостаточность |
• Апноэ во сне |
|
Сердечно-сосудистая система |
• Гипертрофическая кардиомиопатия • Аритмии |
• Поражается редко |
|
Желудочно-кишечный тракт |
• Трудности при кормлении |
• Дисфагия |
|
Органы чувств |
Нейросенсорная тугоухость |
|
Младенческая форма может проявляться внутриутробно, с развитием неиммунной водянки плода и антенатальной гибелью вследствие развития тяжелой сердечной недостаточности [6].
Средний возраст манифестации симптомов около двух месяцев [2]. Первыми признакам чаще всего являются генерализованная быстропрогрессирующая мышечная гипотония и слабость - симптом вялого ребенка. Отмечается задержка или регресс моторного развития. Несмотря на мышечную слабость, наблюдается псевдогипертрофия, преимущественно икроножных мышц, которые при пальпации обычно имеют плотную консистенцию [2]. Часто возникают трудности с кормлением в виде дисфагии из-за гипотонии лица, слабости мышц глотки, гортани и языка, макроглоссии. Поражение диафрагмы и дыхательной мускулатуры приводит к дыхательной недостаточности (ДН). По мере прогрессирования ДН дети нуждаются в мерах респираторной поддержки в виде аппаратов CPAP, BiPAP, искусственной вентиляции легких, установке трахеостомы [6]. Для предотвращения застойных явлений в легких детям проводится регулярная санация дыхательных путей, постуральный и вибрационный массаж, физиотерапия. Дисфагия в совокупности со слабостью межреберных мышц и диафрагмы часто приводит к хроническому аспирационному синдрому и/или аспирационной пневмонии, в связи с чем дети грудного возраста нуждаются в установке назогастрального зонда для профилактики развития данных осложнений. Явления бронхообструкции не типичны для заболевания [6].
В результате накопления гликогена в миокарде происходит утолщение стенок обоих желудочков, межжелудочковой и межпредсердной перегородок. Со временем формируется симметричная гипертрофическая кардиомиопатия с обструкцией выводных отделов и развитием жизнеугрожающей сердечной недостаточности. На поздних стадиях гипертрофия может трансформироваться в дилатационную кардиомиопатию [6]. Увеличение размеров сердца также приводит к уменьшению объема легких, сдавлению бронхов и прогрессированию дыхательной недостаточности.
У детей с БП может присутствовать гепатомегалия, обычно обусловленная сердечной недостаточностью. Возможно развитие нейросенсорной потери слуха и необходимость использования слуховых аппаратов.
Вследствие разрушения миоцитов в биохимическом анализе крови может отмечаться повышение креатинфосфокиназы (КФК), аспартатаминотрансферазы (АСТ), аланинаминотрансферазы (АЛТ), лактатдегидрогеназы (ЛДГ) [3]. На электрокардиограмме (ЭКГ) регистрируется нарушение проведения и бивентрикулярная гипертрофия в виде укорочения интервала PRu увеличения вольтажа комплексов QRS [6]. На рентгенограмме обнаруживается кардиомегалия и увеличение кардио-торакального индекса (КТИ) выше возрастной нормы.
При отсутствии терапии дети погибают в возрасте до 2 лет жизни от сердечно-легочной недостаточности и инфекционных осложнений [2, 7].
Поздняя (взрослая) форма манифестирует в любом возрасте старше 12 мес. Протекает данная форма мягче, чем инфантильная, но в конечном итоге является тяжело инвалидизирующей. Спектр клинических проявлений широко вариабелен. Основными проявлениями являются медленно прогрессирующая проксимальная миопатия конечностей, особенно нижних [2]. Из-за слабости мышц туловища у пациента возникает неуклюжесть при выполнении физических упражнений, лордоз/сколиоз, ригидность позвоночника, походка вперевалку, так называемая «утиная походка»; в дальнейшем исчезает способность к самостоятельному передвижению. Часто возникают дыхательные нарушения, апноэ во сне из-за слабости диафрагмы, которые по мере прогрессирования приводят к необходимости ИВЛ и тяжелым респираторным инфекциям [6]. Каждый год течения заболевания при отсутствии терапии увеличивает на 13% и 8% вероятность использования инвалидной коляски и ИВЛ соответственно. У пациента развивается бульбарный синдром со слабостью языка в виде дизартрии и дисфагии. Также симптомами могут быть недержание мочи и кала, быстрая утомляемость, нейросенсорная тугоухость. Кардиомиопатия и гепатомегалия при данной форме встречаются значительно реже. Возможны-дилатация восходящего отдела аорты, аневризмы крупных артерий в результате артериопатии [8].
Тяжесть заболевания и возраст смерти зависит от остаточного уровня фермента, возраста манифестации, степени поражения органов и миопатии, присоединения интеркуррентных состояний и скорости прогрессирования заболевания [3].
Диагноз БП устанавливается на основании энзимодиагностики «методом сухих пятен крови» с определением активности а-1,4-глюкозидазы [3]. Полный дефицит фермента <1% связан с инфантильной формой БП, частичная активности уровня 2-40% приводит к поздней форме БП [7]. Подтверждается диагноз при обнаружении мутации гена GAA методом молекулярно-генетического тестирования. Однако необходимо учитывать наличие мутаций с неопределенным значением и вариантов псевдодефицита. В сложных диагностических случаях или при псевдодефиците активности фермента может помочь определение одного из биомаркеров тетрасахарида глюкозы в моче.
Электронейромиография и биопсия мышц являются неспецифическими и непоказательными методами исследования у пациентов с болезнью Помпе [3]. Поэтому их использование рекомендуется для дифференциальной диагностики миопатических синдромов. Во многих странах мира выявление дефицита а-1,4-глюкозидазы входит в программу неонатального скрининга, так как более ранняя диагностика и, как следствие, ранний старт терапии снижают заболеваемость и смертность. Пренатальная диагностика возможна с помощью анализа ДНК при наличии родственников с установленным диагнозом БП [2].
Дифференциальная диагностика проводится с большим перечнем заболеваний (табл. 3).
Таблица 3. Дифференциальный диагноз болезни Помпе
Форма БП | Этиология | Сходные черты | Отличительные черты |
Инфантильная форма | |||
Спинальная мышечная атрофия 1 (болезнь Верднига -Гоффмана) | Ген: SMN1 Тип наследования: аутосомно-рецессивный | Гипотония, трудности при кормлении, прогрессирующая слабость проксимальных мышц и арефлексия | Отсутствие кардиомиопатии |
Болезнь Данона | Ген: LAMP2 Тип наследования: Х-сцепленный | Гипотония, гипертрофическая кардиомиопатия, миопатией | Умственная отсталость |
Нарушение поглощения карнитина (OMIM 212140 ) | Ген: SLC22A5 Тип наследования: аутосомно-рецессивный | Мышечная слабость, кардиомиопатия | Не характерно повышение концентрации КК в сыворотке. Может наблюдаться энцефалопатия или кома |
Поздняя форма БП | |||
Конечностно-поясная мышечная дистрофия | Ген: Тип наследования: аутосомно-рецессивный | Прогрессирующая мышечная слабость наблюдается в ногах, тазе и плечах | Нет поражения мышц туловища |
Мышечная дистрофия Дюшенна - Беккера | Ген: Тип наследования: Х-сцеплен-ный | Прогрессирующая слабость проксимальных мышц, дыхательная недостаточность и затруднения при передвижении | Почти не встречается у женщин |
Другие гликогенозы | Ген: Тип наследования: Х-сцепленный | Отсутствие гипогликемии при БП | |
Полимиозит | Прогрессирующая, симметричная мышечная слабость | ||
ТЕРАПИЯ
В настоящее время единственным патогенетическим методом лечения является ферментзаместительная терапия, разработанная методом генной инженерии. В 2006 г. была одобрена терапия рекомбинантной человеческой а-1,4-глюкозидазой (rhGAA) [3, 5]. Препарат вводится медленно внутривенно в дозировке 20 мг/кг 1 раз в 2 недели. На фоне терапии замедляется прогрессирование заболевания, восстанавливается сердечная и скелетная мускулатура, происходит обратное развитие кардиомиопатии, повышается выживаемость и качество жизни пациентов, отдаляется время необходимости ИВЛ [9].
Однако на результат терапии влияет статус перекрестно-реактивного иммунологического материала (CRIM). У пациентов с CRIM-отрицательным статусом нулевая экспрессия GAA, в связи с чем у них раньше проявляются антитела к rhGAA в высоких титрах, ответ на терапию становится хуже, снижается выживаемость [5]. Для уменьшения этой реакции пациентам проводится комплексная иммуносупрессивная терапия [7]. В базе данных вариантов GAA есть данные о возможном CRIM статусе при наличии определенной мутации [5]. Эту информацию можно использовать в клинической практике для определения прогноза и тактики терапии в условиях отсутствия возможности диагностики уровня титра антител.
В августе 2021 г. FDA одобрило новый препарат для лечения БП с поздним началом - авалглюкозидаза альфа. Данный препарат в 15 раз больше имеет точек сродства с рецептором маннозо-6-фосфата, ключевым путем транспортировки фермента GAA в лизосому [10]. Клиническую эффективность препарат показал на всех этапах исследования [11]. Дозировка препарата зависит от веса тела, 20 мг/кг каждые 2 недели для пациентов весом 30 кг и более, а для пациентов с массой менее 30 кг рекомендуется доза 40 мг/кг каждые 2 недели [10].
Продолжаются исследования в области генной терапии, такие как использование векторов для доставки гена фермента в клетки. Генная терапия может в перспективе стать более эффективным методом лечения, направленным на коррекцию генетического дефекта непосредственно на уровне ДНК пациента [3].
В стационаре ДГКБ им. З.А. Башляевой диагноз инфантильной формы болезни Помпе был поставлен четырем детям. Ниже приведен клинический пример одного из них, при котором имело место сочетание врожденного порока сердца и инфантильной формы болезни Помпе.
КЛИНИЧЕСКИЙ СЛУЧАЙ
Мальчик Е., 06.04.2022 г. р. Ребенок от матери 33 лет с отягощенным соматическим анамнезом (аутоиммунный тиреоидит в стадии компенсации, хронический пиелонефрит), второй беременности, протекавшей на фоне угрозы прерывания, ОРВИ, первых срочных, самостоятельных родов. Масса тела при рождении 3630 г, длина тела 53 см, оценка по шкале Апгар 8/9 баллов.
Через два часа после рождения отмечена отрицательная динамика в виде нарастания дыхательной недостаточности, церебральной депрессии; выслушан систолический шум. Ребенок переведен в ОРИТН родильного дома, где ему проводилась респираторная поддержка в режиме nCPAP в течение 9 часов, инфузионная, антибактериальная терапия с положительной динамикой. На вторые сутки жизни (07.04.2022) проведена ЭХОКГ в связи с подозрением на врожденный порок сердца (ВПС), обнаружены дефект межжелудочковой перегородки (ДМЖП), дефект межпредсердной перегородки (ДМПП).
На третьи сутки жизни (08.04.2022) ребенок переведен в неонатологическое отделение № 1 ДГКБ им. З.А. Башляевой г. Москвы.
По данным ЭХОКГ, подтвержден ВПС (ДМЖП перимембранозный 8-9 мм, частично прикрыт аневризмой, толщина миокарда (ТМ) 6 мм (норма 4,5-5мм), градиент систолического давления (ГСД) между желудочками (ЛЖ/ ПЖ) 18 мм рт. ст., межпредсердное сообщение (МПС) 5 мм. Рентгенография органов грудной клетки: расширение тени сердца, гиперволемия в малом круге кровообращения.
В связи с клиникой сердечной недостаточности ребенку была назначена терапия: дигоксин в дозе 0,00001 г/кг/сут, спиронолактон 4 мг/кг/сут, калия аспаргинат и магния аспаргинат. На 17-е сутки (22.04.2022) с положительной динамикой, нарастанием ГСД ЛЖ/ПЖ до 40 мм рт. ст., ребенок выписан домой.
В 2 мес (04-11.06.2022) при динамическом обследовании в кардиологическом отделении ДГКБ им. З.А. Башляевой отмечены дефицит массы тела 13%, одышка при нагрузке, субиктеричность лица и склер, двустороннее гидроцеле.
Лабораторно: гипербилирубинемия (билирубин 113 мк-моль/лзасчетнепрямойфракции),гиперферментемия(АЛТ 143 Ед/л, АСТ 252 Ед/л, щелочная фосфатаза 1443,6 Ед/л (норма до 450 Ед/л), КФК 619,5 Ед/л (норма до 295 Ед/л)). Ультразвуковое исследование печени: размеры печени не увеличены, перегиб желчного пузыря.
Проведена коррекция базисной кардиологической терапии. В связи с наличием синдрома цитолиза, холестаза, перегиба желчного пузыря назначен курс урсодезоксихолиевой кислоты. Рекомендован динамический контроль измененных биохимических параметров, с особым вниманием на параметр КФК.
В 2,5 мес по месту жительства отмечено нарастание желтухи, сохранение гиперферментемии, нарастание КФК до 677 Ед/л. Учитывая вышеизложенное, кровь ре-бенка направлена на энзимодиагностику методом тандемной масс-спектрометрии с целью исключения лизосомальных болезней накопления, спинальной мышечной атрофии, врожденной мышечной дистрофии Дюшенна, болезни Помпе. 29.07.2022 получены данные о резком снижении активности а-1,4-глюкозидазы до 0,21 мкМ/л/ч (норма 1-25), что характерно для БП. Дальнейшее молекулярно-генетическое исследование выявило компа-унд-гетерозиготное состояние в гене GAA с.1082С>Т/р. Pro361Leu, с.1425del/p.Leu476Ter, подтверждающее диагноз болезни Помпе.
В 3 мес (24.07.2022-05.08.2022) мальчик повторно госпитализирован в кардиологическое отделении ДГКБ им. З.А. Башляевой. При осмотре обращало на себя внимание появление клинических симптомов, характерных для болезни Помпе: специфический фенотип лица с горизонтальным разрезом глаз и припухлостью век, а также макроглоссия, плотные при пальпации икроножные мышцы, умеренная мышечная гипотония, затруднение при кормлении, одышка при физической нагрузке, дисгармоничность физического развития за счет дефицита массы тела (19%), задержка темпов моторного развития.
ЭКГ: признаки бивентрикулярной гипертрофии миокарда. ЭХОКГ: размеры ДМЖП прежние, увеличение среднего давления в легочной артерии до 28 мм рт. ст., гипертрофия миокарда левого и правого желудочков без обструкции выходных отделов.
По жизненным показаниям с 4,5 мес (25.08.2022) мальчик начал получать заместительную ферментотерапию рекомбинантной человеческой а-1,4-глюкозидазой (rhGAA) из расчета 20 мг/кг 1 раз в 2 недели внутривенно, под контролем лабораторных и инструментальных показателей.
На фоне терапии отмечены улучшение аппетита, уменьшение гипертрофии икроножных мышц, макроглоссии. Физическое развитие к году стало гармоничным (рис. 1).
Лабораторно отмечалось снижение и стабилизация показателей КФК, АСТ, АЛТ, ЛДГ, но они оставались повышенными относительно средневозрастной нормы (рис. 2, 3, 4, 5).
После инициации терапии уменьшились и достигли нормы показатели толщины миокарда (толщина задней стенки левого желудочка уменьшилась с 6,7 мм до 5 мм, межжелудочковой перегородки с 7,8 мм до 5,5 мм) (рис. 6, 7). По течению порока сердца также отмечена положительная динамика. Размеры дефекта при последнем обследовании уменьшились до 6 мм (рис. 8), ГСД между желудочками нарастает и нормализовался КДР ЛЖ (рис. 9, 10). Улучшение отмечено и по результатам рентгенографии ОГК с разницей в 1 год (рис. 11).
При динамическом наблюдении нервно-психическое развитие с положительной динамикой (табл. 4).
Таблица 4. Нервно-психическое развитие пациента в процессе динамического наблюдения
|
Возраст |
Количество инфузий препарата |
Моторное развитие |
Речевое развитие и навыки |
|
5 мес |
3 инфузии |
Держал голову, поворачивался со спины на бок |
Улыбался, гулил, мимика лица активная |
|
8 мес |
9 инфузий |
Самостоятельно переворачивался со спины на живот и обратно, сидел при поддержке |
Активный лепет |
|
11 мес |
15 инфузий |
Сидел самостоятельно, стоял с опорой, активно ползал |
|
|
1 год 2 мес |
22 инфузии |
Стоял с опорой, ходил и бегал с поддержкой |
Играл в игрушки, произносил отдельные слоги |
|
1 год 6 мес |
30 инфузий |
Ходил, бегал самостоятельно |
|
|
1 год 10 мес |
39 инфузий |
Произносил отдельные слова, сам пьет из чашки, кушает, частично самостоятельно раздевается |
Рис. 1. Физическое развитие пациента в процессе динамического наблюдения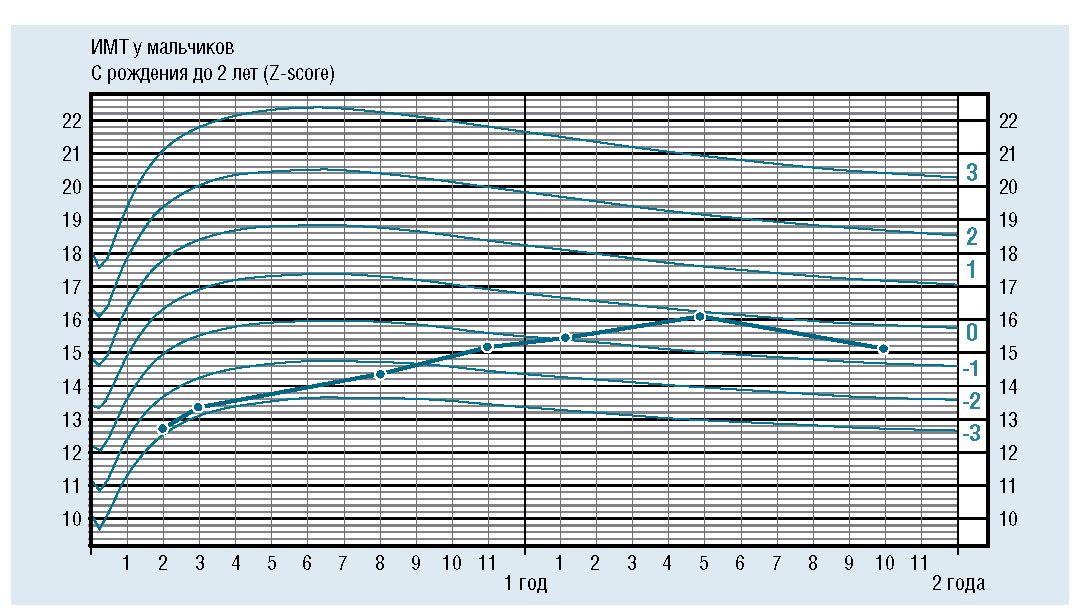
Рис. 2. Динамическое изменение уровня КФК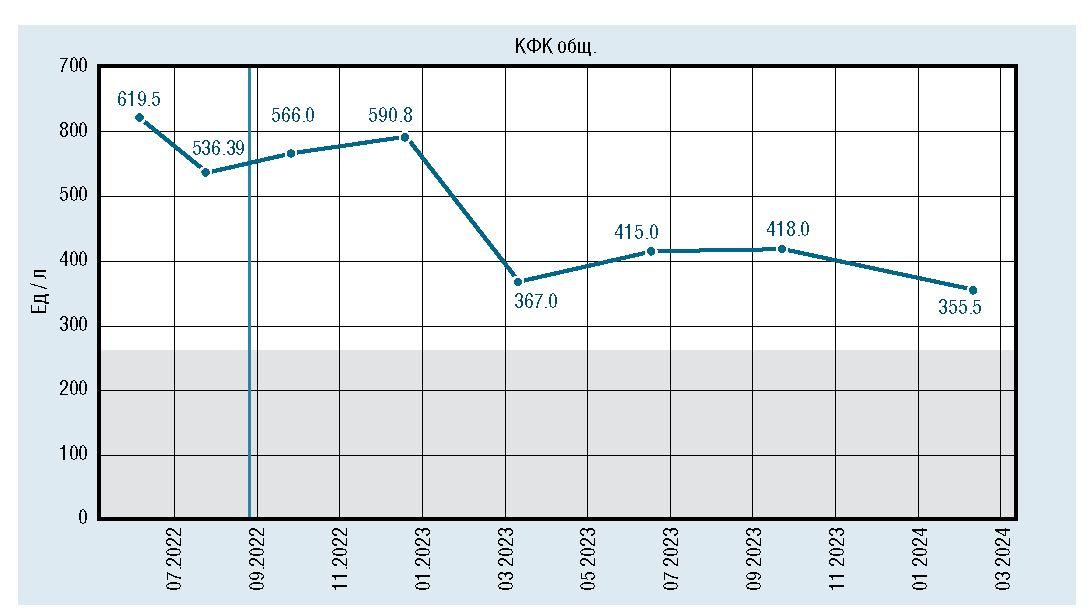
Рис. 3. Динамическое изменение уровня АЛТ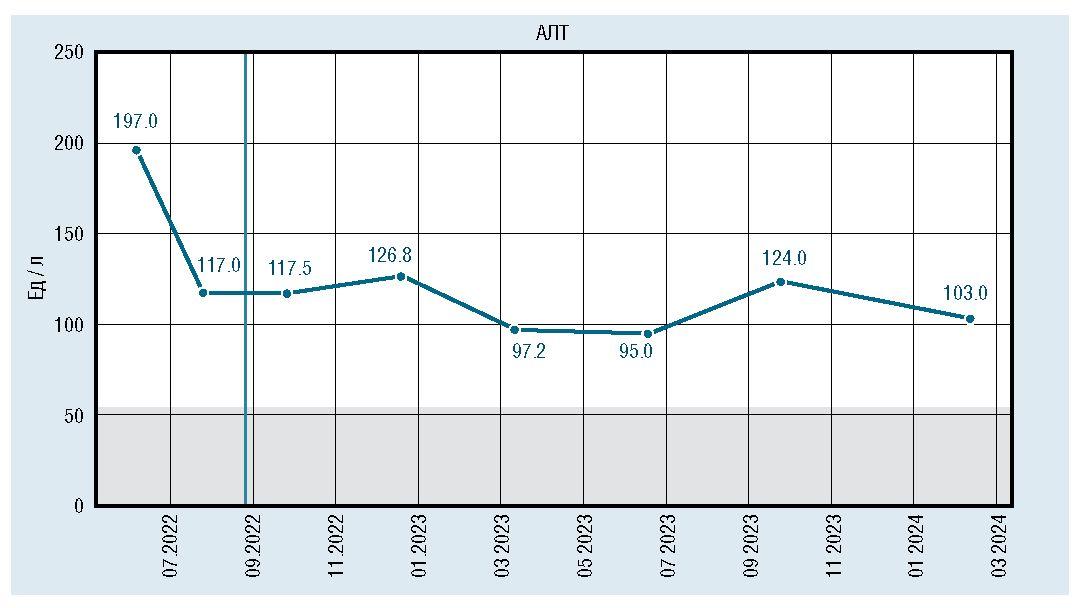

Рис. 4. Динамическое изменение уровня АСТ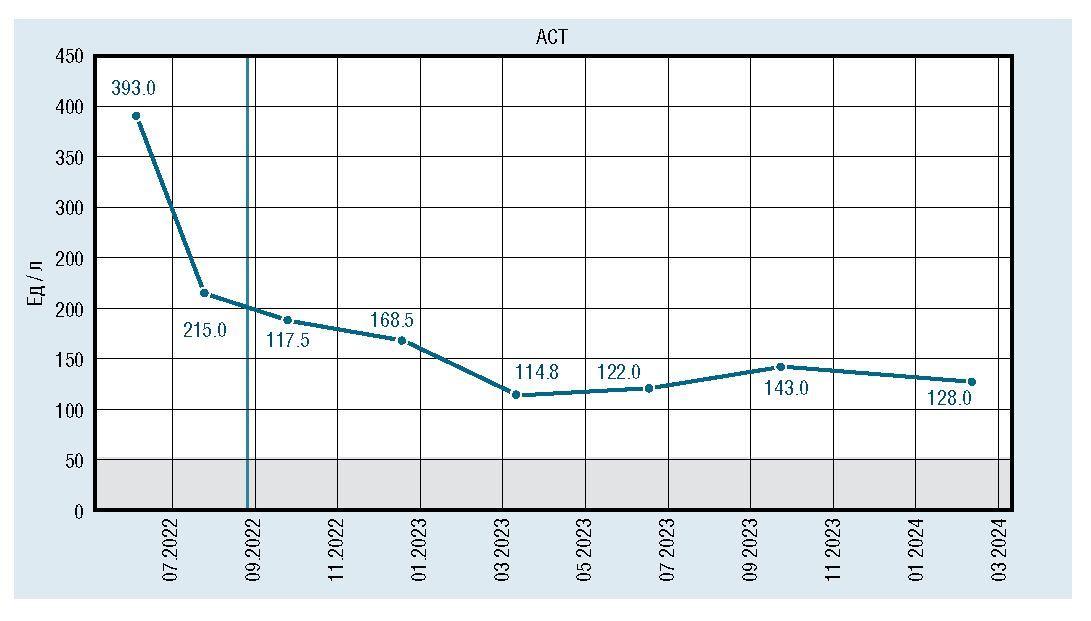

Рис. 5. Динамическое изменение уровня ЛДГ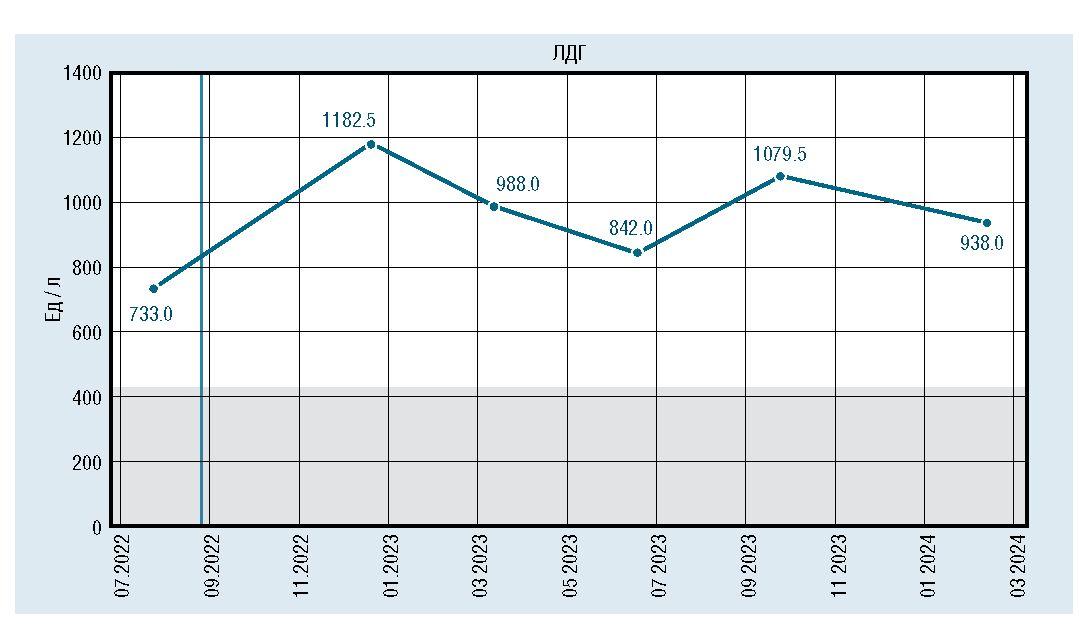
Рис. 6. Динамическое изменение толщины миокарда задней стенки левого желудочка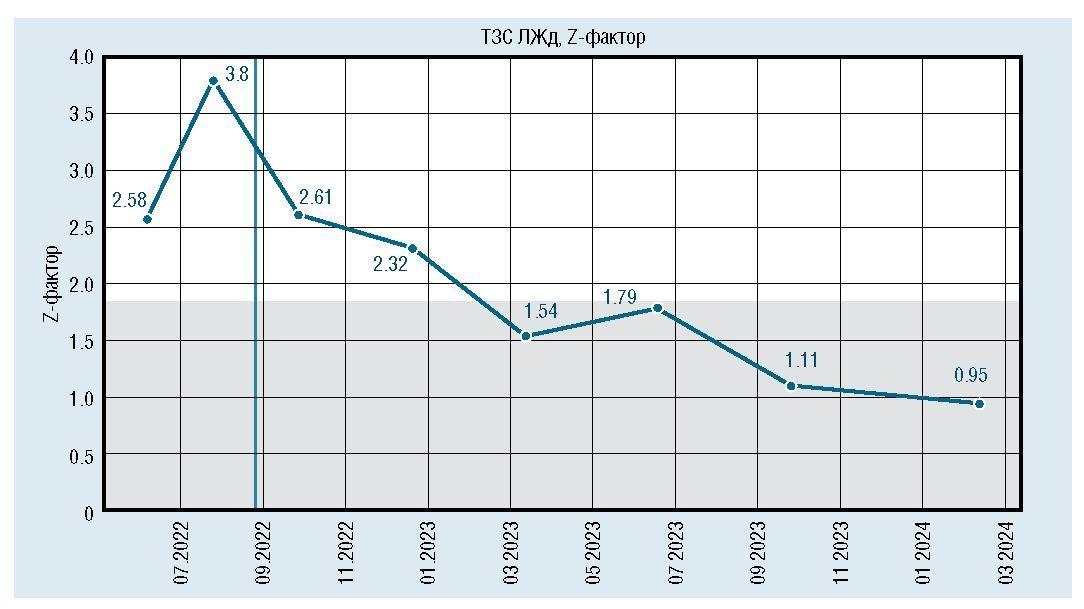
Рис. 7. Динамическое изменение толщины межжелудочковой перегородки в диастолу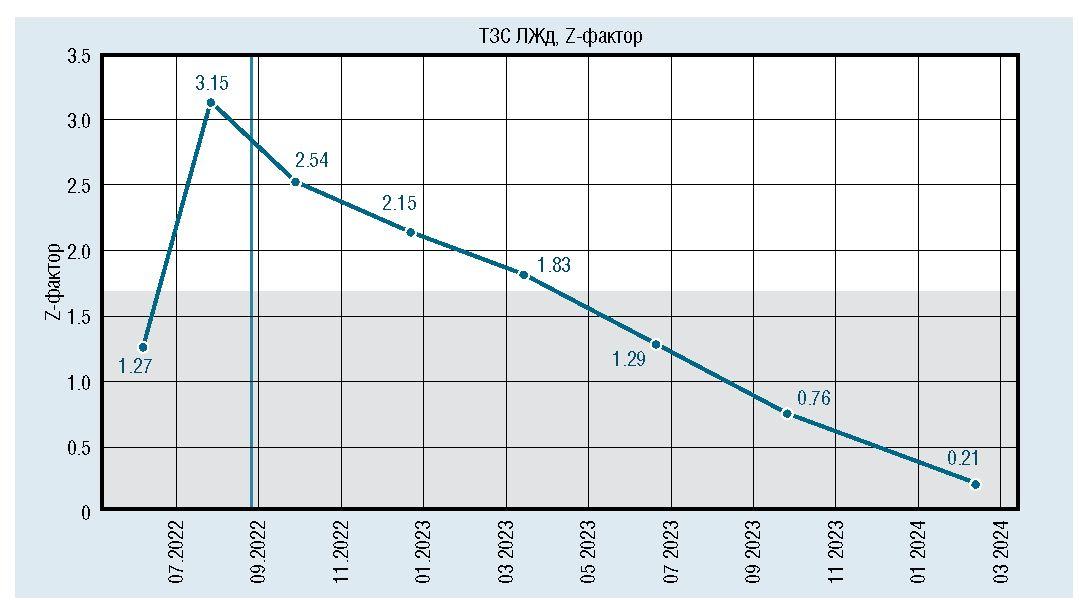

Рис. 8. Динамическое изменение размера дефекта межжелудочковой перегородки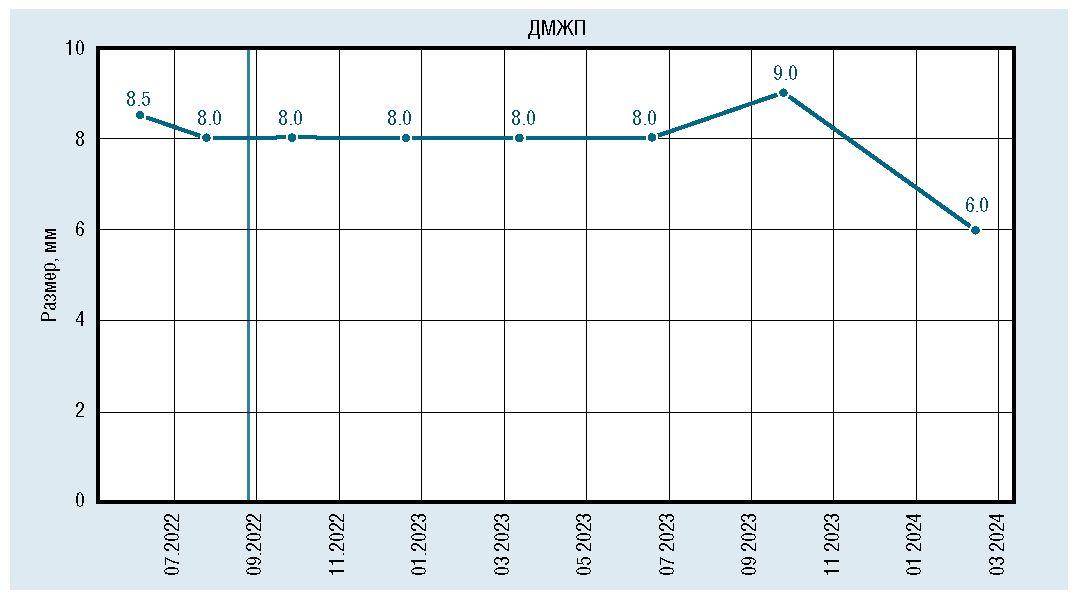

Рис. 9. Динамическое изменение градиента давления между желудочками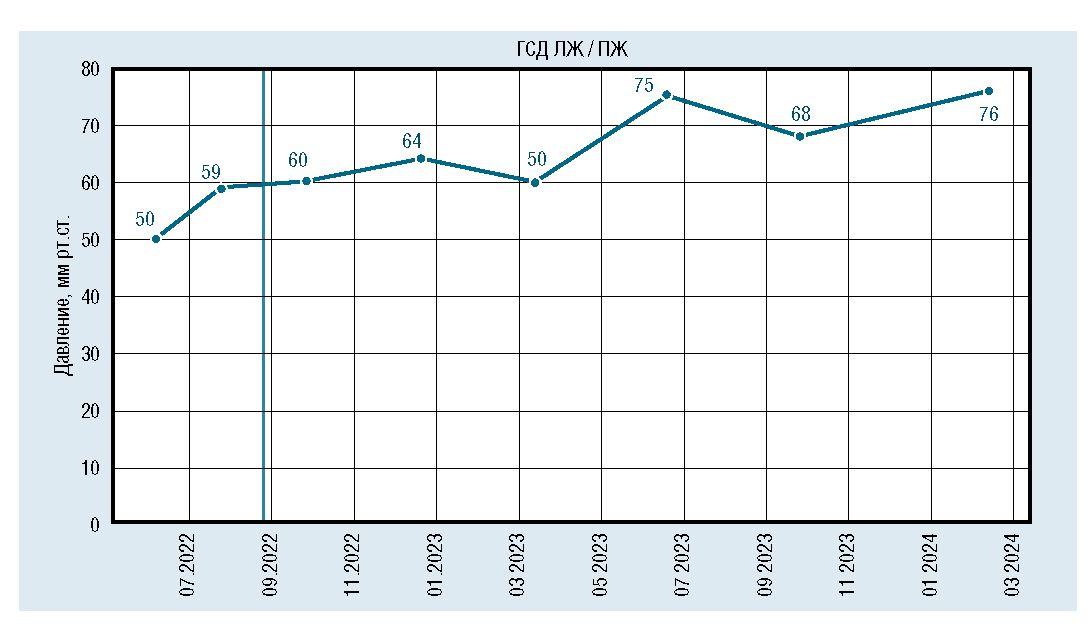
 Рис. 10. Динамическое изменение конечного диастолического размера левого желудочка
Рис. 10. Динамическое изменение конечного диастолического размера левого желудочка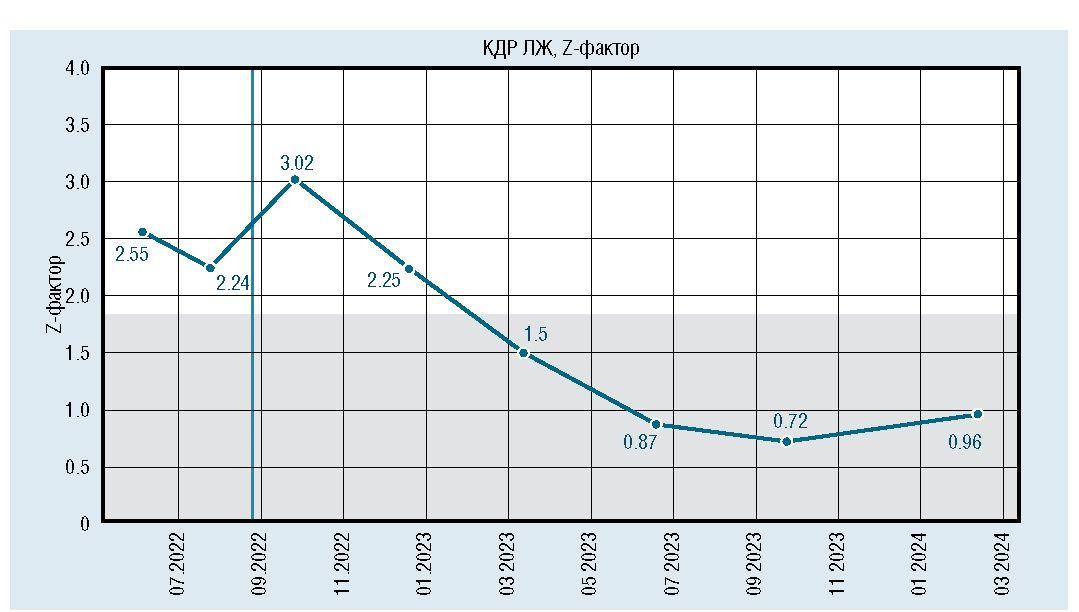
Рис. 11. Рентгенограмма органов грудной клетки ребенка в возрасте 11 мес
(март 2023 года и 1 года 10 мес (февраль 2024 года).
Кардиоторакальный индекс при измерении 57% и 50% соответственно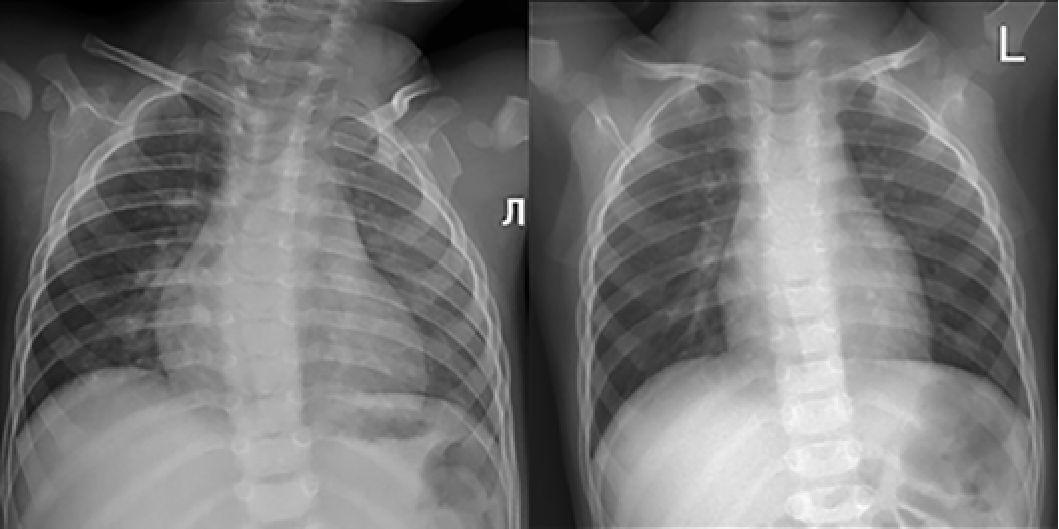
 Проспективное наблюдение пациента продолжается по настоящее время. Каждые 3-4 месяца ребенок госпитализируется в кардиологическое отделение ДГКБ им. З.А. Башляевой для динамического наблюдения и коррекции терапии. На данный момент ему 3 года, он продолжает получать ФЗТ по основному заболеванию (болезнь Помпе) по месту жительства. По ВПС мальчик гемодинамически стабилен и в настоящее время не нуждается в проведении кардиохирургической коррекции ВПС. ГСД ЛЖ/ПЖ без отрицательной динамики. Минимизирована терапия сердечной недостаточности.
Проспективное наблюдение пациента продолжается по настоящее время. Каждые 3-4 месяца ребенок госпитализируется в кардиологическое отделение ДГКБ им. З.А. Башляевой для динамического наблюдения и коррекции терапии. На данный момент ему 3 года, он продолжает получать ФЗТ по основному заболеванию (болезнь Помпе) по месту жительства. По ВПС мальчик гемодинамически стабилен и в настоящее время не нуждается в проведении кардиохирургической коррекции ВПС. ГСД ЛЖ/ПЖ без отрицательной динамики. Минимизирована терапия сердечной недостаточности.
ОБСУЖДЕНИЕ
Особенность представленного клинического случая заключается в том, что у ребенка с ВПС при плановом кардиологическом обследовании в возрасте 2 мес было выявлено изменение лабораторных показателей в виде гиперферментемии. Это позволило заподозрить инфантильную форму БП до появления симптомов заболевания, своевременно верифицировать данный диагноз и начать лечение. По данным мировой литературы, дети с БП имеют более высокий риск развития осложнений при хирургическом вмешательстве, в связи с чем у данного ребенка на фоне стабильной гемодинамики оперативное лечение ВПС решено было отложить на более поздний возраст. Несмотря на сочетание у пациента двух тяжелых коморбидных патологий, раннее начало комбинированной терапии (ФЗТ и терапии СН) позволяет на данном этапе откладывать кардиохирургическую коррекцию, с улучшением течения основного заболевания и хорошей динамикой физического, психического, речевого и стато-моторного развития.
ЗАКЛЮЧЕНИЕ
Болезнь Помпе, являющаяся редким генетическим заболеванием, требует комплексного подхода в диагностике, лечении и уходе за пациентами. В данной статье были рассмотрены различные аспекты этого заболевания, включая его патогенетические механизмы, формы заболевания, клиническую картину, методы диагностики и новые подходы к терапии.
Представленный клинический случай ранней диагностики и успешного лечения пациента подчеркивает важность своевременного выявления симптомов, комплексного обследования и назначения эффективной терапии. Междисциплинарный подход, включая совместное участие врачей различных специальностей и координацию усилий, играет значительную роль в управлении сложными случаями таких сочетанных заболеваний.
Благодаря постоянно проводимым научным исследованиям и развитию медицинской практики, продолжают совершенствоваться методы диагностики, лечения и реабилитации для пациентов с болезнью Помпе и сопутствующими заболеваниями. Мы надеемся, что дальнейшие исследования позволят улучшить прогноз жизни этих пациентов и предложат им еще более эффективные терапевтические подходы. Ближайшая перспектива - включение в протокол ведения пациентов с инфантильной формой болезни Помпе препарата авалглюкозидазы-альфа [12]. Он содержит увеличенное количество маннозо-6-фос-фатных остатков в сравнении с алглюкозидазой альфа, что улучшает поглощение фермента клетками-мишенями, приводит к кратному увеличению клиренса гликогена и потенциальному улучшению клинических исходов. В настоящее время, которое считается переходным периодом, осуществляется внедрение данного препарата не только у взрослых пациентов, но и у детей [12, 13]. Происходит переключение с одного препарата на другой. Готовится к такому переходу и наш пациент.
Информация о финансировании: Финансирование данной работы не проводилось.
Конфликты интересов: Авторы заявляют об отсутствии конфликтов интересов.
Литература
1. Peruzzo P., Pavan E., Dardis A. Molecular genetics of Pompe disease: a comprehensive overview // Annals of Translational Medicine. 2019. Vol. 7, No. 13. P. 278. DOI: 10.21037/atm.2019.04.13.
2. Клинические рекомендации «Болезнь Помпе». Одобрено Научно-практическим советом Минздрава РФ, 2019.
3. Pompe disease: pathogenesis, molecular genetics and diagnosis/ S. Taverna[et al.] // Aging (Albany NY). 2020. Vol. 12, No. 15. P. 1585615874. DOI: 10.18632/aging.103794.
4. Colburn R., Lapidus D. An analysis of Pompe newborn screening data: a new prevalence at birth, insight and discussion // Frontiers in Pediatrics. 2024. Vol. 11. P. 1221140. DOI: 10.3389/ fped.2023.1221140.
5. Extension of the Pompe mutation database by linking disease-associated variants to clinical severity / M.Y. Nino [et al.] // Human Mutation. 2019. Vol. 40, No. 11. P. 1954-1967. DOI: 10.1002/humu.23854.
6. Младенческая форма болезни Помпе: клиника, диагностика, лечение / Н.П. Котлукова [и др.] // Нервно-мышечные болезни. 2012. № 4. С. 66-74.
7. Dose-intensive therapy (DIT) for infantile Pompe disease: A pilot study / R. Jeanine[et al.] // Molecular Genetics and Metabolism Reports. 2025. Vol. 42. P. 101179.
8. Living with Pompe disease: results from a qualitative interview study with children and adolescents and their caregivers / Moritz Ilan Truninger[et al.] //Orphanet Journal of Rare Diseases. 2024. Vol. 19. P. 358. DOI: 10.1186/s13023-024-03368-7.
9. Резолюция Совета экспертов «Клинический мониторинг пациентов с болезнью Помпе с поздним началом. Влияние ферментозаместительной терапии нового поколения на клинические исходы у пациентов с болезнью Помпе с поздним началом» // Нервно-мышечные болезни. 2023. Т. 13. №2. С. 83-85.
10. Государственный реестр лекарственных средств. ОХЛП Нексвиазайм ЛП-№(002078)-(РГ-RU) от 31.03.2023. Доступно по: https:// grls.rosminzdrav.ru/Grls_View_v2. aspx?routingGuid=62dc44e5-1640-47fc-befb-c4ae06957cb1.
11. Safety and efficacy of avalglucosidase alfa inindividuals with infantile-onset Pompe disease enrolled in the phase 2, open-label Mini-COMET study:The 6-month primary analysis report / Priya S. Kishnani [et al.] //Genetics in Medicine. 2023. Vol. 25, No. 2. P. 100328. DOI: 10.1016/j.gim.2022.10.010.
12. Safety and efficacy of avalglucosidase alfa versus alglucosidase alfa in patients with lateonset Pompe disease (COMET): a phase 3, randomised, multicentretrial / J. Diaz-Manera [et al.] //Lancet Neurology. 2021. Vol. 20, No. 12. P. 1012-1026.
13. Avalglucosidase alfa in infantile-onset Pompe disease: A snapshot of real-world experience in Italy / Agata Fiumara[et al.] //Molecular Genetics and Metabolism Reports. 2024.Vol. 40. P. 101126.



