Глутаровая ацидурия тип I у детей. Клинические рекомендации.
Статьи
Глутаровая ацидурия тип I у детей
- Союз педиатров России
Оглавление
- Ключевые слова
- Список сокращений
- Термины и определения
- 1. Краткая информация
- 2. Диагностика
- 3. Лечение
- 4. Реабилитация
- 5. Профилактика и диспансерное наблюдение
- 6. Дополнительная информация, влияющая на течение и исход заболевания
- Критерии оценки качества медицинской помощи
- Список литературы
- Приложение А1. Состав рабочей группы
- Приложение А2. Методология разработки клинических рекомендаций
- Приложение А3. Связанные документы
- Приложение Б. Алгоритмы ведения пациента
- Приложение В. Информация для пациентов
- Приложение Г.
Ключевые слова
-
Ацилкарнитин
-
Глутаровая ацидемия
-
Глутаровая ацидурия
-
Диета с ограничением лизина и триптофана
-
Левокарнитин или L-карнитин
-
Рибофлавин
-
Рибофлавин-чувствительная форма глутаровой ацидурии
Список сокращений
3-OH-глутаровая кислота - 3-гидроксиглутаровая кислота
GA - глутаровая кислота
GCDH - фермент глутарил-КоА дегидрогеназа
Good clinical practice (GСP) - надлежащая клиническая практика
Good practice points (GPPs) - индикаторы доброкачественной практики
OMIM - ((Online Mendelian Inheritance in Man) интернет база заболеваний человека, наследуемых по законам Менделя.
T1W - Т1 взвешенное изображение
T2W - Т2 взвешенное изображение
ГА1 - глутаровая ацидурия тип 1
ГАМК - гамма-аминомасляная кислота
КТ - компьютерная томография
МРТ - магнитно-резонансная томография
С5DC – глутарилкарнитин
ТМС - тандемная масс спектрометрия
ЦСЖ - цереброспинальная жидкость
Термины и определения
«Энцефалитоподобный криз» или «метаболический криз»- критическое, угрожающее жизни состояние, спровоцированное неблагоприятными факторами, обуславливающими усиление процессов клеточного катаболизма и проявляющееся угнетением сознания до сопора и комы в результате развивающегося отека и набухания мозга.
1. Краткая информация
1.1 Определение
Глутаровая ацидурия тип 1 (недостаточность глутарил-КоА дегидрогеназы, глутаровая ацидемия тип 1) - аутосомно-рецессивное заболевание, обусловленное мутациями в гене, кодирующем фермент глутарил-КоА дегидрогеназу (GCDH). Дефицит данного фермента приводит к накоплению в биологических жидкостях и тканях глутаровой и 3-OH-глутаровой (3-гидрокси-глутаровой), кислот оказывающих нейротоксическое действие преимущественно на подкорковые структуры головного мозга. Диагноз глутаровой ацидурии тип 1 не является клиническим, он устанавливается при обнаружении определенных биохимических и/или молекулярно-генетических изменений, выявляемых у пациентов с определенной клинической симптоматикой или на доклинической стадии при проведении массового скрининга новорожденных [1,2,5].
1.2 Этиология и патогенез
Ген GCDH картирован на хромосоме 19p13.2. К настоящему времени в гене GCDH описано более 200 различных мутаций, одной из самых распространенных является R402W, встречающаяся с частотой 12 - 40% в странах западной Европы. Некоторые мутации характерны для определенных этнических групп и изолятов: мутации P248L и E365K в Турции, мутации IVS 1 +5G>T и A421V в общинах амишей [2,5,15].
Фермент глутарил-КоА-дегидрогеназа (GCDH) участвует в метаболизме аминокислот лизина, гидролизина, триптофана. GCDH локализована в матриксе митохондрий и представляет собой гомотетрамер. Каждая субъединица этого белка состоит из 438 аминокислот, N-терминальный остаток (44 аминокислоты) удаляется после транспортировки фермента в митохондрию. GCDH катализирует 2 последовательных реакции превращения глутарил-КоА в кротонил-КоА (реакции дегидрогенирования и декарбоксилирования). В результате блокирования данной ферментной реакции происходит накопление глутаровой и 3-OH-глутаровой кислот в биологических жидкостях и тканях ( рис.1) [1,2].

Рис.1 - Схема метаболических процессов, приводящих к развитию глутаровой ацидурии тип: метаболические блоки выделены голубым цветом, патологические метаболиты выделены двойной рамкой
Мутантный фермент относится к группе флавопротеиновых дегидрогеназ митохондриального матрикса, участвующих в переносе электронов к убихинону через электрон-переносящий флавопротеин в дыхательной цепи митохондрий.
Механизмы развития ГА 1 до конца не изучены. Преимущественное поражение стриарной системы связывают с избирательной токсичностью глутаровой кислоты и/или ее производных. Также глутаровая кислота и ее метаболиты ингибируют декарбоксилазу глутаминовой кислоты (фермент, участвующий в метаболизме ГАМК), что вызывает снижение концентрации этого тормозного нейромедиатора. У больных с ГА 1 выявлено значительное снижение декарбоксилазной активности в лобных отделах коры головного мозга, хвостатых ядрах и скорлупе и снижение концентрации ГАМК в цереброспинальной жидкости (ЦСЖ). Однако данные биохимические нарушения могут быть вторичными и возникать вследствие повреждений ГАМК–эргических нейронов мозга.
Механизмы патогенеза острых «энцефалитоподобных» кризов окончательно не ясны. Считают, что глутаровая и 3-OH-глутаровая кислоты, имеющие структурное сходство с глутаматом, взаимодействуют с N-метил-аспартатными рецепторами, для которых глутамат является естественным активатором, что вызывает чрезмерное накопление ионов Са2+ в постсинаптических нейронах и приводит к гибели клеток. Другим возможным нейротоксическим фактором считают накопление промежуточного метаболита обмена триптофана и лизина – квинолиновой кислоты. В настоящее время, до конца не распознана причина лобно-височной атрофии и/или гипоплазии и субдуральных кровоизлияний при ГА1. Считается, что во время эмбриогенеза 3-ОН-глутаровая кислота может нарушать формирование стенок сосудов, в первую очередь – сосудов головного мозга, приводя к повышению их проницаемости и возникновению кровоизлияний [2,14].
При аутопсии у всех пациентов с ГА1 выявляют выраженную субкортикальную и кортикальную атрофию головного мозга, атрофическую вентрикуломегалию. В большинстве случаев визуализируются некротические изменения в области скорлупы и головки хвостатого ядра. Реже поражаются зрительные тракты, мозолистое тело, внутренняя капсула, глубокие отделы белого вещества мозжечка, ствола мозга. У ряда пациентов обнаруживают «губчатую» дегенерацию белого вещества, преимущественно в перивентрикулярных областях, реже в таламусе, бледном шаре и стволе головного мозга [2,14].
У большинства пациентов при аутопсии печени выявляется жировая инфильтрация клеток печени, проксимальных канальцев почек и миокардиоцитов, что может быть связано с неспецифической митохондриальной дисфункцией [2,14].
1.3 Эпидемиология
Глутаровая ацидурия тип 1 (ГА1) относится к числу редких наследственных болезней обмена веществ. Частота заболевания в странах Западной Европы составляет в среднем 1:50 000 живых новорожденных. Высокая частота встречаемости ГА1 отмечена среди общин амишей в Америке, Канаде и Пенсильвании - 1:300. К настоящему времени в литературе имеются описания более 400 случаев этого заболевания [1,2,5].
1.4 Кодирование по МКБ-10
E72.3 - Нарушения обмена лизина и гидроксилизина
1.5 Классификация
По клиническому течению классифицируют два основных подтипа ГА1: с острым/«энцефалитоподобным» и подострым/доброкачественным течением [2].
1.6.Приемеры формулировки диагноза
-
Глутаровая ацидурия тип I
-
Глутаровая ацидемия тип I
-
Нарушения обмена лизина и гидроксилизина
-
Глютарикацидурия, гидроксилизинемия, гиперлизинемия
2. Диагностика
Диагностика основана на данных анамнеза родословной, анамнеза заболевания, клинических симптомах, результатах лабораторных и инструментальных исследований, данных молекулярной ( ДНК) диагностики.
2.1 Жалобы и анамнез
ГА 1 обычно дебютирует в раннем детском возрасте - от 3 до 36 месяцев, с пиком манифестации от 6 до 18 месяцев. В 75% случаев наблюдается «энцефалитоподобный» вариант заболевания. У 75-80% больных макроцефалия может быть первым симптомом болезни, которая отмечается с рождения или развивается в первые месяцы жизни. Нередко, «провоцирующими» факторами манифестации заболевания являются черепно-мозговая травма, хирургические вмешательства, инфекции или вакцинация. В большинстве случаев, заболевание начинается остро, спустя 24-72 часа от воздействия «провоцирующего» фактора. При развитии «энцефалитоподобного» криза практически все больные поступают с жалобами на внезапно развившуюся лихорадку, частые срыгивания или неукротимую рвоту, кишечные расстройства, эпилептические приступы.
У 25 % больных возможны задержка психомоторного развития с постепенной утратой ранее приобретенных навыков, присоединением гиперкинезов, что характеризует более доброкачественное течение заболевания.
2.2 Физикальное обследование
Макроцефалия может быть первым симптомом болезни, которая отмечается с рождения или развивается в первые месяцы жизни.
При развитии «энцефалитоподобного» криза часто происходит угнетение сознания до сопора и комы, в результате развивающегося отека и набухания мозга, нередко приводящего к летальному исходу в первые сутки заболевания. Спустя несколько дней или недель после криза могут отмечаться различные типы гиперкинезов (орофациальные, хореиформные, хореоатетоидные, баллистические), диффузная или односторонняя мышечная дистония, которая часто сочетается со спастичностью. Заболевание носит волнообразный характер: после перенесенных «энцефалитоподобных» кризов происходит медленное, но не полное восстановление неврологических нарушений. В последующем у пациентов с грубым дистоническим синдромом часто формируются вторичные осложнения, такие как гипотрофия, хронический аспирационный синдром, подвывихи суставов и выраженный болевой синдром [6,12].
В 25% случаев заболевание имеет подострое, доброкачественное течение. На первом году жизни дети наблюдаются с задержкой психомоторного развития, а в дальнейшем происходит постепенная утрата ранее приобретенных навыков, и присоединяются различные виды гиперкинезов. Дистонические гиперкинезы приводят к нарушению ходьбы, письма и речевых функций. Многие больные длительное время наблюдаются у неврологов со спастико-гиперкинетической формой детского церебрального паралича. Другими частыми симптомами являются профузное потоотделение, эпизоды немотивированной лихорадки, эпилептические приступы.
Среди офтальмологических симптомов нередко обнаруживают кровоизлияния в сетчатку, катаракту, офтальмопарез, косоглазие и пигментную дегенерацию сетчатки. У некоторых пациентов наблюдаются атактические расстройства, очень редко встречаются миоклонии и тремор. У большинства пациентов интеллект не страдает или наблюдаются легкие когнитивные нарушения. У не леченных больных смертельный исход наступает в течение первого десятилетия жизни на фоне тяжелого метаболического криза или в результате развития Рейе-подобного синдрома.
У ряда больных с ГА1 на первом году жизни в 10-30% случаев возникают субдуральные кровоизлияния, как правило, после перенесенной легкой черепно-мозговой травмы. Пациенты с субдуральными гематомами и/или битемпоральными арахноидальными кистами в сочетании с макроцефалией и дистониями должны быть отправлены в генетическую лабораторию для исключения ГА1. Диагноз глутаровой ацидурии тип I должен быть обязательно исключен лабораторными методами у пациентов с предполагаемым синдромом «тряски младенца» («shake syndrome») [2,9].
Клинический диагноз ГА1 может быть заподозрен у пациентов, имеющих макроцефалию, экстрапирамидные нарушения с дебютом в раннем детском возрасте и характерные нейрорадиологические изменения [8]. В таблице представлены критерии, позволяющие предположить ГА1 (табл. 1).
Таблица 1 - Клинико-диагностические критерии, позволяющие заподозрить глутаровую ацидурию тип 1 [16].
|
Вероятность диагноза |
Низкая |
Умеренная |
Высокая |
|
Клинические критерии |
-Макроцефалия; -Выступающие лобные бугры; -Нарушения вскармливания -Задержка двигательного развития |
-Рейе-подобные эпизоды (острая метаболическая энцефалопатия) -энцефалитоподобные эпизоды, -гиперкинетическая форма детского церебрального паралича. -периодическая атаксия |
-Острая энцефалопатия -прогрессирующие экстрапирамидные нарушения -орофациальные дискинезии |
|
Семейный анамнез |
Не отягощен
|
Синдром «внезапной смерти младенца» или смертельный исход от некупируемых судорог в семье |
Наличие в семье больного с подтвержденным диагнозом ГА тип 1, наличие смертельного исхода в семье от острого симметричного некроза базальных ганглиев |
|
Нейрорадио- логические критерии |
-атрофия/гипоплазия лобных отделов -задержка миелинизации -вентрикуломегалия -расширение субарахноидальных пространств |
-Субдуральные скопления -атрофия/гипоплазия височных отделов -лейкоэнцефалопатия (подростки, взрослые) -изолированное поражение бледного шара |
-Лобно-височная атрофия головного мозга, -изолированные симметричные некрозы базальных ганглиев (бледного шара и хвостатых ядер) |
2.3 Лабораторная диагностика
Основными методами подтверждающей диагностики ГА1 являются биохимические и нейрорадиологические методы исследования.
-
Рекомендуется определение в биологических жидкостях (кровь, моча) органических кислот и ацилкарнитинов (глутарилкарнитина) методом тандемной масс-спектрометрии [2,7] и или хроматомасс спектрометрии [4,14].
(Сила рекомендации C; уровень убедительности доказательств II)
Комментарии: С помощью тандемной масс спектрометрии (ТМС) определяют концентрации глутарилкарнитина (С5DC) в пятнах высушенной крови. При данном заболевании концентрации глутаровой, 3-ОН-глутаровой кислот и глутарилкарнитина повышаются в десятки раз по сравнению с нормой.
У некоторых пациентов с ГА1 концентрация глутаровой кислоты в моче и крови может быть в пределах нормы, в таких случаях диагностически-значимым маркером является повышение концентрации 3-OH-глутаровой кислоты, что и позволяет установить диагноз ГА1 [2,4,7,14,19,20].
-
Рекомендуется определение активности глутарил-КоА дегидрогеназы (в лейкоцитах и фибробластах кожи) [2,5,14,20].
(Сила рекомендации C; уровень убедительности доказательств II)
-
Рекомендуется молекулярная ДНК диагностика (выявление мутаций в гене GCDH) [7,14,17,20].
(Сила рекомендации C; уровень убедительности доказательств II)
Комментарии: Определение активности фермента и ДНК диагностика необходимы как для верификации диагноза, так и для медико-генетического консультирования семей, последующей пренатальной диагностики.
-
Рекомендуется мониторирование уровня органических кислот в крови и моче [1,2,7,17, 19,20].
(Сила рекомендации C; уровень убедительности доказательств II)
Комментарии: Токсическое повреждение головного мозга вследствие высоких концентраций глутаровой и 3-OH-глутаровой кислот делает необходимым регулярное проведение исследования содержания данных метаболитов в биологических средах. Снижение концентрации глутаровой и 3-OH-глутаровой кислот в моче наблюдается у пациентов, находящихся на специфической низкобелковой диете, а также при терапии рибофлавином у больных с рибофлавин-чувствительными формами. Однако концентрации данных кислот в сыворотке крови и ЦСЖ на лечении не снижаются до нормы.
2.4 Инструментальная диагностика
-
Рекомендуется использовать нейрорадиологические методы: компьютерная томография (КТ), магнитно-резонансная томография (МРТ) [7,11,13,19,20].
(Сила рекомендации C; уровень убедительности доказательств II)
Комментарии: Наиболее частыми нейрорадиологическими признаками при ГА1 являются: лобно-теменная гипоплазия/атрофия, вентрикуломегалия, субдуральные гематомы, задержка миелинизации/демиелинизация и некроз базальных ганглиев. Эти изменения могут встречаться как изолированно, так и в любом сочетании. Характерной нейрорадиологической особенностью ГА 1 является симметричное расширение сильвиевых щелей с формированием «эффекта надкушенного яблока» или «крыльев летучей мыши», которая встречается у 95%. У большинства не леченых пациентов выявлено расширение субарахноидальных пространств даже на пресимптоматической стадии заболевания, которые не претерпевают никаких изменений при проведении контрольных МРТ головного мозга и сочетаются с нормальной толщиной коры головного мозга и нормально развитыми извилинами.
У большинства пациентов субдуральные гигромы/гематомы, часто располагаются билатерально, реже - имеют одностороннюю локализацию. Описывают образование битемпоральных арахноидальных или субэпендимарных кист, редко встречается их односторонняя локализация. Изменения белого вещества головного мозга встречаются у многих больных с ГА 1 и часто сочетаются с некрозом базальных ганглиев. Поражение белого вещества, как правило, симметричное, преимущественно в перивентрикулярных зонах в области передних и задних рогов боковых желудочков и в области семиовального центра, в некоторых случаях обнаруживают демиелинизацию субкортикальных U-волокон. Остается не ясным, являются ли данные области очагами демиелинизации или гипомиелинизации. У некоторых больных описано поражение мозолистого тела [13, 16]. Характерные нейрорадиологические изменения при ГА 1 приведены в Приложении Г1.
2.5 Иная диагностика
-
Рекомендуются консультации специалистов (по показаниям) генетика, офтальмолога, невропатолога, диетолога, кардиолога, логопеда-дефектолога, нейроортопеда пациентам с глутаровой ацидурией типI, имеющим нарушения функций соотвествующих органов и систем [1,2,7].
(Сила рекомендации С; уровень убедительности доказательств II)
2.6. Дифференциальная диагностика
-
Все описанные выше клинические, лабораторные и нейрорадиологические признаки заболевания не считаются патогномоничными, при их наличии заболевание следует заподозрить. Подтверждается ГА 1 путем ферментативного анализа и/или обнаружением двух клинически значимых (патогенных) мутаций, при этом рекомендуется пользоваться имеющимися базами данных, например, OMIM [20, 23].
(Сила рекомендации C; уровень убедительности доказательств II)
3. Лечение
Стратегия лечения направлена на коррекцию метаболических нарушений комплексом мероприятий: консервативными методами (патогенетичксая диетотерапия, левокарнитин, рибофлавин), хирургическими методами (лечение лечение внутричерепных кровоизлияний и костных деформаций), реабилитационными метолами. Лечение должны быть начато всем больным с выявленным достоверным повышением уровня глутаровой кислоты в моче до получения результатов ДНК диагностики ГА1. Для лечения «энцефалитоподобного» криза в международные стандарты включены методы интенсивной терапии. Также проводится симптоматическая терапия с тщательным подбором лекарственных препаратов, направленных на коррекцию неврологических нарушений.
3.1 Консервативное лечение
-
Рекомендуется патогенетическая диетотерапия терапия с исключением высокобелковых продуктов, богатых лизином и триптофаном, обязательныи тспользованием специализированных продуктов на основе смесей аминокислот без указанных патогенетически значимых аминокислот [1,3,7,18,1,20].
(Сила рекомендации В; уровень убедительности доказательств II)
Комментарии: Диетические рекомендации подбираются пациентам индивидуально. Диетотерапия при заболевании направлена на снижение поступления лизина, триптофана, которые являются основными предшественниками нейротоксических метаболитов - глутаровой и 3-OH-глутаровой кислот. Данные аминокислоты содержатся в большом количестве в белках животного происхождения (рыбе, мясе, молочных продуктах и др.). В среднем содержание в них лизина составляет 6 - 9% и триптофана 0,6-2%. В растительных же продуктах (овощи, фрукты) содержание указанных аминокислот минимально.
При ГА 1 применяют два варианта лечебного питания: первый - диета с низким содержанием лизина и использованием специализированного продукта, который является более предпочтительным, особенно для детей раннего, младшего и среднего возраста, так как при нем специализированный продукт (табл. 2) компенсирует дефицит белка, второй вариант - диета с низким содержанием общего белка
Таблица 2 - Специализированные продукты на основе аминокислот без лизина и триптофана*
|
Наименование продукта |
Белок (экв.) г |
Жир, г |
Углеводы, г |
Энергети ческая ценность, ккал |
Возраст применения |
|
«Нутриген 14 -trp,- lys» |
14 |
23 |
50,4 |
472 |
от 0 до 1 года |
|
«Нутриген 40 --trp,- lys» |
40 |
13 |
32,2 |
406 |
старше 1 года |
|
«Нутриген 70 -trp,- lys» |
70 |
0 |
5,8 |
303 |
старше 1 года |
|
MTVI Anamix GA1 |
13,1 |
23 |
49,8 |
466 |
от 0 до 1 года |
|
XLYS,TRY Глуаридон |
79 |
- |
4 |
332 |
старше 1 года |
*-продукты зарегистрированы на территории Российской Федерации
Применение второго варианта диеты повышает риск алиментарной недостаточности незаменимых аминокислот таких как триптофан, что может привести к развитию различных нарушений: повышенной возбудимости, нарушению сна, немотивированным колебаниям температуры, пеллагре (в результате снижения образования никотиновой кислоты), поэтому данный вариант может быть применен только у пациентов старшего возраста. Минимально допустимые потребности патогенетически значимых аминокислот в питании детей различного возраста представлены в таблице 3 [1,2,18].
Таблица 3 - Ориентировочная потребность в лизине и триптофане у больных глутаровой ацидурией тип 1 в зависимости от возраста
|
Возраст больных |
Суточная потребность в аминокислотах мг/кг массы тела |
|
|
Лизин |
Триптофан |
|
|
0 - 3 мес |
80-100 |
10-20 |
|
3 - 6 мес |
70-90 |
10-15 |
|
6 - 9 мес |
60-80 |
10-12 |
|
9 - 12 мес |
50-70 |
10-12 |
|
1 – 4 года |
55-65 |
8-12 |
|
4 – 7 лет |
45-55 |
7-11 |
|
7 – 11 лет |
35-45 |
4-10 |
|
Девушки 11- 15 лет |
30-40 |
4-6 |
|
Девушки 15- 18 лет |
20-30 |
3-5 |
|
Юноши 11- 15 лет |
30-40 |
4-6 |
|
Юноши 15- 18 лет |
35-45 |
6-8 |
Пациенты младенческого возраста могут находиться на грудном вскармливании. Грудное молоко в данном случае будет являться основным источником натурального белка в сочетании со специализированной смесью аминокислот, соответствующей возрасту ребенка. Количество лизина в грудном молоке известно и составляет 86 мг на 100 мл. Диетотерапия с низким содержанием лизина и триптофана должна строго соблюдаться у пациентов до 6 лет.
Если после шестилетнего возраста у пациентов не возникают «энцефалитоподобные» кризы, возможно использование низкобелковой диеты без специализированного продукта, однако в этом случае необходим контроль за нутритивным статусом ребенка, чтобы вовремя предотвратить развитие белково-энергетической недостаточности и дефицитных состояний. Специализированная диета требует мониторинга за уровнем аминокислот и их метаболитов в плазме крови и моче, биохимических маркеров нутритивного статуса (общий белок, протеинограмма и др.), динамического наблюдения за физическим развитием и нутритивным статусом пациента, а также периодической коррекции лечебного рациона с расчетом его химического состава. При расчете низко-белковой диеты со сниженным содержанием лизина и триптофана необходимо помнить, что лизин и триптофан содержатся практически во всех продуктах.
В приложении Г2 представлены таблицы, в которых содержатся международные и российские рекомендации по суточным нормам потребления белка и данные по среднему содержанию лизина в некоторых натуральных продуктах.
Медикаментозная терапия.
-
Рекомендуется пожизненно патогенетическая терапия левокарнитином (L- карнитин) [1,2,7,16,19,20].
(Сила рекомендации В; уровень убедительности доказательств II)
Комментарии: L- карнитин связывает глутаровую кислоту и обеспечивает ее выведение из организма в виде глутарилкарнитина. У большинства не леченых пациентов с ГА 1 концентрация общего и свободного карнитина в плазме и сыворотке крови низкая. Начальная доза L- карнитина составляет 100 мг/кг/сутки, она может быть снижена до 50 мг/кг/сутки у детей старше 6 лет.
-
Рекомендуется по показаниям лечение рибофлавином [7, 10, 20].
(Сила рекомендации С; уровень убедительности доказательств II)
Комментарии: Рибофлавин-чувствительная форма ГА1 встречается крайне редко. Однако, у пациентов с данной формой заболевания при назначении рибофлавина в дозе 100-200 мг в сутки отмечается улучшение состояния в виде уменьшения степени выраженности неврологических нарушений. При улучшении клинического и метаболического статусов у больного ребенка при назначении рибофлавина необходимо продолжить его применение, при отсутствии таковых препарат должен быть отменен.
В Приложении Г3 суммированы основные мероприятия, которые обязательно должны быть выполнены после подтверждения диагноза ГАI.
3.2 Хирургическое лечение
-
Рекомендуется по показаниям хирургическое лечение субдуральных гидром и арахноидальных кист [7,19,20].
(Сила рекомендации С; уровень убедительности доказательств II)
Комментарии: Решение о целесообразности проведения нейрохирургического вмешательства (вентрикулоперитонеальное шунтирование и эвакуация субдуральных скоплений в мозге при прогрессирующей макроцефалии) принимается в каждом конкретном случае консилиумом врачей специалистов. Нейрохирургическая терапия арахноидальных кист и субдуральных гематом у пациентов с ГА1 должна проводиться крайне осторожно под наблюдением детского нейрохирурга.
-
Рекомендуется по показаниям хирургическое лечение костных деформаций (тяжелые контрактуры, вывих бедра и др.) [2,7].
(Сила рекомендации С; уровень убедительности доказательств II)
Комментарии: Решение о целесообразности проведения нейроортопедического вмешательства принимается в каждом конкретном случае консилиумом врачей специалистов.
3.3 Лечение в период метаболического криза
Необходимо помнить, что острый «энцефалитоподобный» криз может развиться при любом инфекционном заболевании, вакцинации или хирургических вмешательствах в основном в возрасте до шести лет. Настораживающими симптомами, свидетельствующими о развитии декомпенсации по основному заболеванию являются: повторные рвоты, диарея, неврологические расстройства (мышечная гипотония/ ригидность, повышенная возбудимость, мышечная дистония, угнетение сознания). У детей старше 6 лет риск развития острых неврологических нарушений во время кризов снижается, что связано с повышением толерантности мозга к колебаниям кислотно-щелочного состояния и воздействию токсических метаболитов.
-
Рекомендуется в период метаболического криза интенсивная терапия, коррекцию патогенетической диетотерапии и доз карнитина [7,19,20].
(Сила рекомендации В; уровень убедительности доказательств II)
Комментарии: Неотложная терапия должна начинаться немедленно и продолжаться весь период фебрильной лихорадки, после вакцинации, до и после хирургического вмешательства в возрасте до 6 лет.
Основными принципами лечения во время «энцефалитоподобных» состояний являются:
-
восстановление катаболического статуса, путем назначения высокоэнергетической инфузионной терапии (глюкоза в сочетании с инсулином);
-
ограничение образования органических кислот, путем снижения поступления белка натуральных продуктов или полное их исключение, по возможности в течение 24-72 часов используют только специализированные продукты на основе смесей аминокислот;
-
усиление детоксикационных механизмов организма и предотвращение вторичного истощения собственного пула карнитина, путем дополнительного введения L-карнитина;
-
восстановление кислотно-основного равновесия организма.
-
Рекомендуется интенсивная инфузионная терапия в стационаре в период метаболического криза [7,19,20].
(Сила рекомендации А; уровень убедительности доказательств II)
Комментарии: Интенсивную терапию необходимо начинать незамедлительно во время заболеваний, протекающих с фебрильной лихорадкой, после вакцинации и хирургических вмешательств. Раннее начало интенсивного лечения позволяет предотвратить развитие тяжелых неврологических нарушений у большинства пациентов с глутаровой ацидурией 1 типа.
При появлении у больного повторных рвот, повышения температуры 38,50С и более, неврологических расстройств показана срочная госпитализация в стационар. Основные принципы интенсивной терапии в условиях стационара представлены в таблице 4.
Таблица 4 - Основные принципы терапии в условиях стационара [7,19,20]
|
А. Восстановление энергетической потребности |
||||
|
Калории |
Повышение энергетической потребности до 120% от возрастной нормы |
|||
|
120% (расчет диеты на ккал/кг/день)1 |
0-6 месяцев |
7-12 месяцев |
1-3 года |
4-6лет |
|
98-128 |
96-109 |
98-109 |
96-98 |
|
|
Б. Внутривенная инфузионная терапия |
||||
|
Декстрозаж 20% |
15-20 г/кг/день |
|||
|
Жиры 20% |
Начальная доза 1-2 г/кг/день, если возможно, повышение дозы до 2-3 г/кг/день |
|||
|
Коррекция электролитных нарушений |
Электролиты крови необходимо поддерживать на верхней границе нормальных значений |
|||
|
Инсулино терапия |
При транзиторной гипергликемии выше 150 мг/дл и/или глюкозурии подключают инсулинж,вк – стартовая доза 0,05 МЕ инсулина/кг/час (контролировать уровень калия в крови) |
|||
|
L-карнитин |
100-200 мг/кг/день |
|||
|
Рибофлавин |
150 мг/день |
|||
|
С. Потребность в белке |
||||
|
Натуральный белок |
Полное исключение белка максимально на 24-48 часов, в дальнейшем с постепенным введением в течение 3-4 дней. Если ребенок находится на низко-белковой диете без приема аминокислотных смесей, осуществляют постепенное введение белка в течение 1-2 дней. |
|||
|
Аминокислотные смеси (без лизина) |
Аминокислотные смеси назначают per os или через назогастральный зонд в дозе 0,8-1,3 г/кг/день |
|||
|
Д. Фармакотерапия |
||||
|
Жаропонижаю- щие средства |
При повышение температуры выше 38,5С – ибупрофенж,вк в дозе 10-15 мг/кг/день2 |
|||
|
Антибактериаль- ная терапия |
Назначают в зависимости от предполагаемого патогенного агента в возрастных дозах |
|||
|
Противорвотные средства |
Метоклопрамид ж,вк в дозе 0,1 мг/кг (не более 3 введений в сутки) |
|||
|
Дегидротацион- ная терапия |
Если диурез менее, чем 3-4 мл/кг/день, назначают фуросемид ж,вк в дозе 0,5-1,0 мг/кг (не более чем, 3-4 инъекции в день) (следить за электролитными нарушениями в крови) |
|||
|
Бикарбонат натрия |
При выраженном ацидозе |
|||
|
Антиэпилепти- ческая терапия |
При судорогах назначают фенобарбитал ж,вк или фенитоин ж,вк в возрастных дозах |
|||
|
Е. Контроль жизненно-важных функций и лабораторных показателей |
||||
|
Жизненно-важные функции |
Частота сердечных сокращений, частота дыхательных движений, контроль артериального давления, температуры, диуреза. При угнетении сознания- оценка уровня сознания по шкале Глазго. |
|||
|
Кровь |
Глюкоза крови, показатели кислотно-основного равновесия, кальций, фосфор, общий анализ крови, креатинин, мочевина, С реактивный белок, аминокислоты крови, карнитиновый статус, креатинфосфокиназа, липаза/амилаза, магний. |
|||
|
Моча |
Кетоновые тела, pH мочи, органические кислоты |
1-во время «энцефалитоподобных» кризов необходимо увеличивать поступление энергии до 120% от нормальной энергетической потребности в сутки, с целью предупреждения развития неврологических нарушений, во время острых состояний энергетическая потребность возрастает на 30-40% от физиологической нормы;
2- у пациентов с ГА1 при лихорадке опасно применение парацетамола, в связи с дополнительного истощением запаса глутатиона в организме, являющегося естественным антиоксидантом.
-
Рекомендуется обязательное наблюдение невропатолога для оценки тяжести неврологических нарушений и их последующей коррекции [2,7,16,19,20].
(Сила рекомендации С; уровень убедительности доказательств II)
Комментарии: Частыми неврологическими нарушениями являются:
-
экстрапирамидные нарушения (более чем в 95% случаев, развиваются после перенесенного острого токсико-метаболического криза);
-
эпилептические приступы (4-40%);
-
субдуральные кровоизлияния (10-30%);
-
высокая вероятность развития двусторонних арахноидальных кист.
-
Рекомендуется фармакотерапия экстрапирамидных нарушений препаратами из группы бензодиазепинов [2,16].
(Сила рекомендации С; уровень убедительности доказательств II)
Комментарии: Препаратами первой очереди для коррекции мышечной дистонии является баклофенж и препараты из группы бензодиазепинов (диазепамж, клоназепамж), направленные на коррекцию генерализованных и фокальных дистоний. Доза препаратов подбираются индивидуально согласно общепринятым стандартам терапии неврологических нарушений. Интратекальное применение баклофена может быть использовано при выраженных дистониях и спастичности.
Препаратом второй очереди при выраженном дистоническом синдроме считается препарат Тригексифенидилж, который назначается детям в возрасте старше 5 лет. Применение антихолинергических препаратов следует ограничивать в связи с риском появления или усугублении когнитивных и психических нарушений.
-
Рекомендуется ботуллинотерапия по показаниям [7,16,20].
(Сила рекомендации С; уровень убедительности доказательств II)
Комметарии: Альтернативным методом лечения тяжелых фокальных дистоний и спастичности является ботулинический токсин типа А ж,вк. Однако, ее следует применять строго по показаниям и с осторожностью, так как при длительном применении возможно формирование тяжелой аутоиммунной реакции.
-
Рекомендуется избирательно по показаниям назначение амантадина ж (трициклический адамантанамин) в возрастных дозировках [2,20].
(Сила рекомендации С; уровень убедительности доказательств II)
-
Рекомендуется терапия эпилептических приступов с индивидуальным подбором противосудорожных препаратов [7,16,19,20].
(Сила рекомендации С; уровень убедительности доказательств II)
Комментарии: Эпилептические приступы возникают у небольшого числа пациентов во время или после перенесенных «энцефалитоподобных» кризов, могут быть однократными и не повторяться в дальнейшем. В ряде случаев за эпилептические приступы врачи ошибочно принимают грубые гиперкинезы. При наличии судорог препаратами выбора являются: фенобарбитал, фенитоин, карбамазепин, топирамат и ламотриджин.
-
Не рекомендуется назначать препараты солей вальпроевой кислоты [2,7,16,20].
(Сила рекомендации С; уровень убедительности доказательств II)
Комментарии: вальпроаты выводятся из организма в виде вальпроилкарнитина, что истощает резерв свободного карнитина и угнетает работу дыхательной цепи митохондрий.
4. Реабилитация
- Рекомендуются санаторно-курортное лечение и реабилитационные мероприятия в случае развития неврологических нарушений по типу детского церебрального паралич, поражения зрительного анализатора, мероприятия, направленные на улучшение когнитивных и речевых функций, психологопедагогическая корекция.
5. Профилактика и диспансерное наблюдение
Профилактические меры включают медико-генетическое консультирование и пренатальную диагностику, которая проводится с помощью молекулярно-генетического исследования биоптата хориона с выявлением мутации соответствующего гена [2,15].
Неонатальный скрининг в Российской Федерации не проводится.
Пациентам с глутаровой ацидурией тип I часто попадают в отделение патологии детей раннего возраста, неврологическое, инфекционное отделения или реанимацию в состоянии энцефалоподобного криза (первичного или повторного). Продолжительность госпитализации зависит от скорости верификации диагноза, коррекции метаболических нарушений путем проведения интенсивной терапии и начала патогенетической диетотерапии, а также от сроков появления положительной динамики со стороны центральной нервной системы и других органов, скорости восстановления биохимических показателей и в среднем составляет 21 день.
После выписки из стационара ребенок должен находиться на этапе амбулаторно-поликлинического наблюдения у педиатра, невропатолога, генетика, больные с симптоматической эпилепсией нуждаются в наблюдении эпилептолога. Консультации других специалистов (офтальмолог, ортопед, врач ЛФК и др.) назначаются по показаниям.
Ежемесячно необходимо проводить коррекцию лечебного питания и симптоматической терапии. У пациентов, соблюдающих специфическую диетотерапию с целью профилактики недостаточности питания необходимо определять концентрацию аминокислот в крови, в том числе и триптофана.
Всем больным показано определение уровня свободного карнитина и ацилкарнитинов в плазме крови или в пятне высушенной крови методом тандемной масс-спектрометрии (ТМС) для выявления вторичной карнитиновой недостаточности. Необходимо отметить, что уровень ацилкарнитинов может повышаться при назначении L-карнитина.
Показано проведение нейрорадиологического исследования головного мозга (МРТ/КТ) при установлении диагноза, а также после перенесенного «энцефалитоподобного» криза.
Всем больным необходим регулярный контроль общего анализа крови, биохимического анализа крови с исследованием уровня альбумина, кальция, фосфора, печеночных трансаминаз (таблица 5).
Таблица 5 - Биохимический и инструментальный мониторинг [19].
|
Биохимические параметры |
Обоснование назначений |
Частота исследований (возраст) |
||
|
0-12 месяцев |
1-6 лет |
Старше 6 лет |
||
|
Аминокислоты (плазма крови) |
Оценка общего аминокислотного статуса |
Каждые 1-2 месяца |
Каждые 3 месяца |
Каждые 6 -12 месяцев |
|
Уровень триптофана |
Предотвращение резкого снижения триптофана в крови |
Назначают пациентам, находящимся на диете смесями аминокислот, лишенных триптофана и лизина при возникновении проблем с питанием. |
||
|
Уровень карнитина (в плазме или сыворотке крови) |
Профилактика вторичного истощения карнитинового пула, контроль осложнений |
Каждые 1-2 месяца |
Каждые 3 месяца |
Каждые 6-12 месяцев |
|
Общий анализ крови + гемосиндром |
Плановый контроль |
Каждые 6 месяцев |
Каждые 6 месяцев |
Каждые 6 -12 месяцев |
|
Альбумин крови |
Предотвращение резкого снижения белка |
Назначают пациентам, имеющим проблемы с питанием |
||
|
Кальций и фосфор крови |
Профилактика остеопороза, тубулопатии |
Каждые 3 месяца |
Каждые 6 месяцев |
Каждые 12 месяцев |
|
Аспартатаминотрансфераза Аланинаминотрасфераза
|
Плановый контроль, профилактика метаболической декомпенсации |
Каждые 3 месяца |
Каждые 6 месяцев |
Каждые 12 месяцев |
|
Нейрорадиологическое исследование головного мозга (МРТ/КТ) |
Контроль структурных изменений головного мозга |
При установлении диагноза, а также после перенесенного «энцефалитоподобного» криза |
||
Ежегодно пациентам должны проходить углубленную диспансеризацию в условиях дневного стационара (длительность госпитализации не менее 10 суток), где также осуществляются необходимые реабилитационные мероприятия.
Родители должны быть обучены правилам организации терапии в межприступный период и в период угрозы метаболического криза. У родителей ребенка и при ребенке всегда должна быть памятка с указанием неотложных мероприятий в период начинающегося метаболического криза (таблица 6).
Таблица 6 - Мероприятия, направленные на предотвращение развития повторных «энцефалитоподобных» кризов[7,20].
|
Мероприятия |
Содержание мероприятия и методы их реализации |
|
Образование и обучение родителей |
Родители должны быть осведомлены о клиническом течении заболевания и его осложнениях. Они должны быть четко информированы о тактике терапии во время развивающегося «энцефалитоподобного» криза. |
|
Лечебно-просветительская работа |
Необходимо обеспечить в обязательном порядке индивидуальным протоколом основной метаболической и интенсивной инфузионной терапии родителей, врачей детской поликлиники по месту жительства пациента. Родители должны быть обеспечены литературой по клинической картине и лечению ГА1. В лечебном протоколе должен быть указан номер телефона лечащего врача, с которым можно связаться в любое необходимое для пациента время. |
|
Экстренная медицинская помощь на дому |
Родители должны иметь все необходимые лекарственные средства для начала интенсивной терапии на дому. |
|
Обеспечение преемственности в лечении (детская поликлиника, районные больницы, метаболический центр) |
Врачи детских поликлиник/районных больниц по месту жительства пациента, должны быть поставлены в известность о пациентах с ГА1, и инструктированы по тактике лечения. Индивидуальный протокол по лечению пациента должен быть отослан в поликлинику/стационар по месту жительства больного незамедлительно после установления диагноза. Интенсивное лечение должно быть начато до перевода больного в специализированный метаболический центр, хорошо владеющий лечением данной патологии. |
|
Медицинская помощь в выходные, праздничные дни и отпускной период |
При выезде ребенка на отдых (в регионы вдали от дома), необходимо отправить по месту пребывания больного ребенка протокол индивидуального лечения, номер телефона лечащего врача. |
|
Медицинская помощь во время инфекционных заболеваний |
При возникновении инфекционных заболеваний и повышении температуры тела до 38,50С родители пациента как можно скорее, должны поставить в известность о случившемся лечащего врача. |
|
Медицинская помощи при плановых хирургических вмешательствах |
При плановом хирургическом вмешательстве врачи хирурги, анестезиологи должны быть информированы о клиническом течении заболевания, лечащий врач должен отправить в хирургический стационар протокол послеоперационного ведения пациента. Основными рекомендациями являются: избегать голодания, назначение растворов глюкозы и двойной дозы карнитина |
6. Дополнительная информация, влияющая на течение и исход заболевания
Если диетотерапия и применение L-карнитина начато в период новорожденности до развития клинической симптоматики, то у большинства пациентов заболевание протекает бессимптомно. При нарушении рекомендаций по неотложному лечению метаболических кризов заболевание имеет неблагоприятный прогноз с развитием структурных изменений головного мозга и прогрессирующей неврологической симптоматики.
Критерии оценки качества медицинской помощи
Список литературы
-
.Баранов А.А., Намазова-Баранова Л.С., Боровик Т.Э., Ладодо К.С., Бушуева Т.В., Маслова О.И., Кузенкова Л.М., Журкова Н.В., Звонкова Н.Г. и др. Диетотерапия при наследственных болезнях аминокислотного обмена Методическое письмо. Москва. 2013. 97 с.
-
Байдакова Г.В.,Петрухин А.С.,Шехтер О.В.,Захарова Е.Ю.,Ильина Е.С.,Михайлова С.В.,Банин А.В.,Волкова Г.И.,Брюсова И.Б.,Бобылова М.Ю.,Рассказчикова И.В. Глутаровая ацидурия тип 1: клиника, диагностика и лечение. Журнал неврологии и психиатрии им.С.С.Корсакова. 2007.-N 10.-С.4-12
-
MP 2.3.1.2432-08 "Нормы физиологических потребностей в энергии и пищевых веществах для различных групп населения Российской Федерации" (утв. Главным государственным санитарным врачом РФ 18 декабря 2008 г.).
-
Baric I., Wagner P., Feyh P., Liesert M., Buckel W., Hoffmann G.F. Sensitivity and specificity of free and total glutaric acid and 3-hydroxyglutaric acid measurements by stable-isotope dilution assays for the diagnosis of glutaric aciduria type I. //J. Inher. Metab. Dis. ? 1999. ?V. 22. ? P. 867-882
-
Biery B.J., Stein D.E., Morton D.H., Goodman S.I. Gene Structure and Mutations of Glutaryl-Coenzyme A Dehydrogenase: Impaired Association of Enzyme Subunits That is Due to an A421V Substitution Causes Glutaric Acidemia Type I in the Amish. // Am. J. Hum. Genet. ? 1996. ?V. 59. ? P. 1006-1011.
-
Bjugstad K.B., Goodman S.I., Freed C.R. Age at symptom onset predicts severity of motor impairment and clinical outcome of glutaric acidemia type I. //J. Pediatr. ? 2000. ? V.137. ?P. 681-686.
-
Boy N1, M?hlhausen C2, Maier EM3, Heringer J4, Assmann B4, Burgard P4, Dixon M5, Fleissner S3, Greenberg CR6,7, Harting I4,8, Hoffmann GF4, Karall D9, Koeller DM10, Krawinkel MB11, Okun JG4, Opladen T4, Posset R4, Sahm K4, Zschocke J12, K?lker S4; Proposed recommendations for diagnosing and managing individuals with glutaric aciduria type I: second revision. J Inherit Metab Dis. 2016 Nov 16. [Epub ahead of print]
-
Brismar J., and Ozand P.T. CT and MR of the brain in glutaric acidemia type I: a review of 59 published cases and a report of 5 new patients. // AJNR Am. J. Neuroradiol. ?1995. ? V.16. ? P.675-683.
-
Caffey J. The whiplash shaken infant syndrome: manual shaking by the extremities with whiplash-induced intracranial and intraocular bleedings, linked with residual permanent brain damage and mental retardation. //Pediatrics. ? 1974. ? V.54. ? P. 396-403.
-
Chalmers R.A., Bain M.D., Zschocke J. Riboflavin-responsive glutaryl CoA dehydrogenase deficiency. //Mol. Genet. Metab. ?2006. ? V.88. ? P.29-37.
-
Chow C.W., Haan E.A., Goodman S.I., Anderson R.M., Evans W.A., Kleinschmidt-DeMasters B.S., Wise G., McGill J.J., Danks D.M. Neuropathology in glutaric acidaemia type 1. // Acta. Neuropathol. ? 1988. ? V.76. ? P.590-594.
-
Corral I., Martinez C.J.C., Martinez-Pardo M., Gimeno A. Glutaric aciduria type 1: diagnosis in adulthood and phenotypic variability. //Neurologia. ?2001. ?V.16. ? P.377-380.
-
Desai N.K., Runge V.M., Crisp D.E. Magnetic resonance imaging of the brain in glutaric acidemia type I: a review of the literature and a report of four new cases with attention to the basal ganglia and imaging technique. //Invest. Radiol. ? 2003. ?V.38. ? P.489-496.
-
Forstner R., Hoffman G.F., Gassner I., Heideman P., DeKlerk J.B.C., Lawrenz-Wolf B., Doringer E., Weib-Wichert P., Troger J., Colombo J.P., Plochl E. Glutaric aciduria type 1: ultrasonographic demonstrations of early signs. //Pediatr. Radiol. ? 1999. ?V.29. ?P.138-143.
-
Funk C.B., Prasad A.N., Frosk P., Sauer S., Kolker S., Greenberg C.R., Del Bigio M.R. Neuropathological, biochemical and molecular findings in a glutaric acidemia type 1 cohort. //Brain. ? 2005. ?V.128. ? P.711-722.
-
Goodman S.I. Prenatal diagnosis of glutaric acidemias. // Prenat. Diagn. ? 2001. ?V. 21. ? P. 1167-1168.
-
Hoffmann G.F., Athanassopoulos S., Burlina A.B., et all. Clinical Course, Early Diagnosis, Treatment, and Prevention of Disease in Glutaryl-CoA Dehydrogenase Deficiency. // Neuropediatr. ? 1996. ?V. 27. ?P. 115-123.
-
Hoffmann G.F., Zschocke J. Glutaric aciduria type I: From clinical, biochemical and molecular diversity to successful therapy. //J. Inher. Metab. Dis. ?1999. ?V. 22. ?P. 381-
-
Phyllis B. Acosta Nutrition Management of Patient with Inherited Metabolic Disorders-Jones and Bartlett Publishers- 2010 p.476.
-
- S. Kolker, E. Christensen , J. V. Leonard , C. R. Greenberg, B. Burlina et al. Guideline for the diagnosis and management of glutaryl-CoAdehydrogenase deficiency (glutaric aciduria type I) J. Inher. Metab. Dis. - 2007- 30:5–22
-
Stefan K?lker, Ernst Christensen, James V. Leonard ,Cheryl R. Greenberg , Avihu Boneh , Alberto B. Burlina, Alessandro P. Burlina, Marjorie Dixon, Marinus Duran, Angels Garc"a Cazorla, Stephen I. Goodman & David M. Koeller, M?rten Kyllerman,Chris M?hlhausen, Edith M?ller,J?rgen G. Okun , Bridget Wilcken, Georg F. Hoffmann ,Peter Burgard. Diagnosis and management of glutaric aciduria type I – revised recommendations. J Inherit Metab Dis (2011) 34:677–694.
-
World Health Organization (1985): Energy and protein requirements. Report of a Joint FAO/WHO/UNU Expert Consultation(1985). WHO Tech Rep Ser No 724, Geneva.
-
3-Reproduced from Reports of the Scientific Commit for food (thirty first series) 1993.
-
-
Online Mendelian Inheritance in Man/ An Onlaine Catalog of Human Genes and Genetic Disorders Update 23 November 2016 - www.omim.org.
Приложение А1. Состав рабочей группы
-
Баранов А.А. – академик РАН, профессор, д.м.н., Председатель Исполкома Союза педиатров России.
-
Намазова-Баранова Л.С. – академик РАН, проф., д.м.н., заместитель Председателя Исполкома Союза педиатров России.
-
Боровик Т.Э. – д.м.н., проф., член Исполкома Союза педиатров России.
-
Бушуева Т.В. – д.м.н., член Союза педиатров России.
-
Глоба О.В. - к.м.н., член Союза педиатров России.
-
Журкова Н.В. – к.м.н., член Союза педиатров России.
-
Захарова Е.Ю. – д.м.н., проф., член Ассоциации медицинских генетиков.
-
Звонкова Н.Г. - к.м.н., член Союза педиатров России.
-
Кузенкова Л.М. - д.м.н., проф., член Исполкома Союза педиатров России.
-
Куцев С.И. - д.м.н., проф., член-корр. РАН, Президент Ассоциации медицинских генетиков.
-
Михайлова С.В. – д.м.н.
-
Николаева Е.А. - д.м.н., проф., член Ассоциации медицинских генетиков.
-
Новиков П.В. - д.м.н., проф., член Ассоциации медицинских генетиков.
-
Пушков А.А. - к.б.н., член Союза педиатров России.
-
Савостьянов К.В. - к.б.н., член Союза педиатров России.
Конфликт интересов отсутствует.
Приложение А2. Методология разработки клинических рекомендаций
Целевая аудитория данных клинических рекомендаций:
-
педиатры
-
врачи общей семейной практики (семейная медицина)
-
генетики
-
диетологи
-
неврологи
-
медицинские психологи
-
дефектологи
-
студенты медицинских ВУЗов, интерны, ординаторы;
Таблица П1 – Уровни убедительности доказательств
|
Уровень убедительности |
Источник доказательств |
|
I (1) |
Проспективные рандомизированные контролируемые исследования Достаточное количество исследований с достаточной мощностью, с участием большого количества пациентов и получением большого количества данных Крупные мета-анализы Как минимум одно хорошо организованное рандомизированное контролируемое исследование Репрезентативная выборка пациентов |
|
II (2) |
Проспективные с рандомизацией или без исследования с ограниченным количеством данных Несколько исследований с небольшим количеством пациентов Хорошо организованное проспективное исследование когорты Мета-анализы ограничены, но проведены на хорошем уровне Результаты не презентативны в отношении целевой популяции Хорошо организованные исследования «случай-контроль» |
|
III (3) |
Нерандомизированные контролируемые исследования Исследования с недостаточным контролем Рандомизированные клинические исследования с как минимум 1 значительной или как минимум 3 незначительными методологическими ошибками Ретроспективные или наблюдательные исследования Серия клинических наблюдений Противоречивые данные, не позволяющие сформировать окончательную рекомендацию |
|
IV (4) |
Мнение эксперта/данные из отчета экспертной комиссии, экспериментально подтвержденные и теоретически обоснованные |
Таблица П2 – Сила рекомендаций
|
Сила рекомендаций |
Описание |
Расшифровка |
|
A |
Рекомендация основана на высоком уровне доказательности (как минимум 1 убедительная публикация I уровня доказательности, показывающая значительное превосходство пользы над риском) |
Метод/терапия первой линии; либо в сочетании со стандартной методикой/терапией |
|
B |
Рекомендация основана на среднем уровне доказательности (как минимум 1 убедительная публикация II уровня доказательности, показывающая значительное превосходство пользы над риском) |
Метод/терапия второй линии; либо при отказе, противопоказании, или неэффективности стандартной методики/терапии. Рекомендуется мониторирование побочных явлений |
|
C |
Рекомендация основана на слабом уровне доказательности (но как минимум 1 убедительная публикация III уровня доказательности, показывающая значительное превосходство пользы над риском) или нет убедительных данных ни о пользе, ни о риске) |
Нет возражений против данного метода/терапии или нет возражений против продолжения данного метода/терапии Рекомендовано при отказе, противопоказании, или неэффективности стандартной методики/терапии, при условии отсутствия побочных эффектов |
|
D |
Отсутствие убедительных публикаций I, II или III уровня доказательности, показывающих значительное превосходство пользы над риском, либо убедительные публикации I, II или III уровня доказательности, показывающие значительное превосходство риска над пользой |
Не рекомендовано |
Актуализация данных клинических рекомендаций будет проводиться не реже, чем один раз в три года. Принятие решения об обновлении будет принято на основании предложений, представленных медицинскими профессиональными некоммерческими организациями с учётом результатов комплексной оценки лекарственных препаратов, медицинских изделий, а также результатов клинической апробации.
Приложение А3. Связанные документы
-
Приказ Минздравсоцразвития РФ № 185 от 22.03.2006 года «О массовом обследовании новорожденных детей на наследственные заболевания»,
-
Приказ Министерства здравоохранения и социального развития РФ от 16 апреля 2012 г. N 366н "Об утверждении Порядка оказания педиатрической помощи"
-
Приказ Министерства здравоохранения РФ «Об утверждении Порядка оказания медицинской помощи больным с врожденными и (или) наследственными заболеваниями» от 15 ноября 2012 г. N 917н).
-
Постановление Правительства Российской Федерации от 9 апреля 2015 года №333 "Об утверждении Правил формирования перечня специализированных продуктов лечебного питания для детей-инвалидов".
Приложение Б. Алгоритмы ведения пациента
Пациент с подозрением
на глутаровую ацидурию тип I
Диагностика

Консультация профильного специалиста
НЕТ
 ДА
ДА
Мониторинг лабораторных показателей пациента
 ДА
ДА
НЕТ
Интенсивная терапия
Приложение В. Информация для пациентов
Глутаровая ацидурия тип I – наследственное заболевание, обусловленное мутациями в гене, кодирующем фермент глутарил-КоА дегидрогеназу (GCDH). Дефицит данного фермента приводит к накоплению в биологических жидкостях и тканях глутаровой и 3-OH-глутаровой (3-гидрокси-глутаровой), кислот оказывающих нейротоксическое действие. Основными клиническими симптомами заболевания являются прогрессирующие экстрапирамидные нарушения. ГА1 относится к той группе наследственных заболеваний, для которых разработаны методы метаболической коррекции, однако эффективность лечения во многом зависит от сроков установления правильного диагноза и назначения патогенетической терапии.
Поздняя диагностика ГА1, несвоевременная и некорректная терапия приводят к тяжелым и необратимым двигательным нарушениям, а в некоторых случаях, - к гибели ребенка.
Для ГА1 разработаны методы молекулярно-генетической диагностики, что позволяет проводить пренатальную диагностику в семьях с отягощенным анамнезом.
В Российской Федерации диагностика осуществляется путем селективного скрининга при наличии первых симптомов заболевания. Неонатальный скрининг не проводится.
Стратегия лечения направлена на коррекцию метаболических нарушений комплексом мероприятий: консервативными методами (патогенетичксая диетотерапия, левокарнитин, рибофлавин), хирургическими методами при необходимости (лечение внутричерепных кровоизлияний и костных деформаций), реабилитационными методами.
Плановая патогенетическая терапия значительно снижает риск их возникновения, однако не предотвращает их развития. «Энцефалитоподобные» или метаболические кризы обычно развиваются в раннем возрасте, преимущественно до шести лет. Запоздалое лечение метаболического криза имеет серьезные последствия. Поэтому важно знать, что метаболическая декомпенсация может возникнуть во время инфекционных заболеваний, вакцинации, оперативных вмешательств. Тревожными симптомами в таких случаях могут быть: рвота, диарея, повышение температуры, нарастание или появление новых неврологических нарушений.
Интенсивная терапия на дому
При наличии у больного с глутаровой ацидурией тип I фебрильной лихорадки ниже 38,50С и отсутствии таких симптомов как, рвота, отказ от еды и различных неврологических нарушений показано продолжение основной метаболической терапии и максимальное ограничение поступления натурального белка с пищей на срок до 12 часов. В данный период необходим осмотр врача каждые 2 часа с оценкой уровня сознания, неврологического статуса и контроля температуры. При стабильном состоянии пациента на протяжении всего наблюдаемого периода продолжают плановую метаболическую терапию и постепенно вводят натуральный белок до разрешенной нормы в течение 24-48 часов (таблица 1).
Таблица 1 - Основные принципы терапии на дому ( K?lker S. et al. 2007)
|
А. Углеводы |
Мальтодекстрин |
||
|
Возраст |
% |
Ккал/100мл |
Объем жидкости (мл) в день через рот |
|
0-1 |
10 |
40 |
Мин. 150 мл/кг |
|
1-2 |
15 |
60 |
120 мл/кг |
|
2-6 |
20 |
80 |
1200-1500 |
|
Старше 6 |
Интенсивная терапия продолжается в том же объеме, что и в возрасте 0-6 лет, возможна индивидуальная адаптация объема и дозы. |
||
|
|||
|
Белок натуральных продуктов |
Полная отмена белка от 24 до 48 часов, далее с постепенным повышением до необходимого количества белка в сутки |
||
|
Специализированные смеси аминокислот |
Специализированные смеси аминокислот назначают согласно разработанным стандартам лечения заболевания (таблица 2) |
||
|
|||
|
L-Карнитин |
Повышение дозы L карнитина в два раза: 200 мг/мг/сутки через рот |
||
|
Жаропонижающие средства |
При повышении температуры тела свыше 38,50С показано назначение жаропонижающих средств, в том числе, ибупрофен, в дозе 10-15 мг/кг/сутки, максимальное назначение препарата до 3-4 раз в сутки и не более чем 60 мг/сутки. |
Приложение Г.
Приложение Г1. Нейрорадиологические изменения (МРТ) головного мозга у пациентов с ГА типI
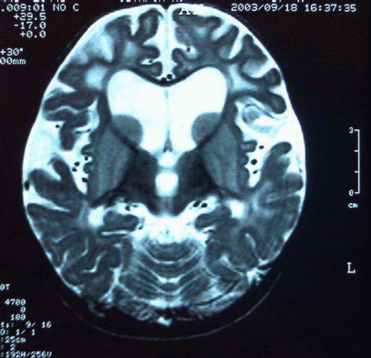
Рисунок А. Эффект «надкушенного яблока» или «крыльев летучей мыши» [2].
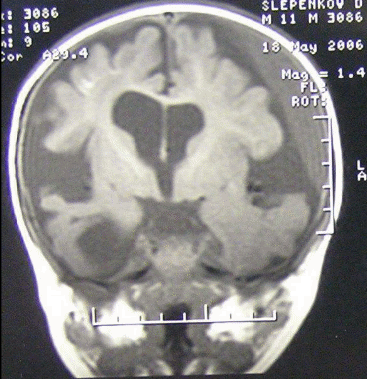

Рисунок В . Субдуральные гематомы [2].
Приложение Г2. Рекомендуемые нормы потребления белка и среднее содержание лизина в различных пищевых продуктах (справочная информация для составления лечебного рациона пациентам с ГА тип I)
Таблица 1 - Рекомендуемые нормы потребления белка для здоровых детей различного возраста [1,3,18,22]
|
Возраст |
RDA США1 (г/кг/сутки) |
ВОЗ2 (г/кг/сутки) |
Евросоюз3 (г/кг/сутки) |
Россия4
|
|
0-6 мес |
2,2 |
1,38 |
- |
2,2 (г/кг/сутки) |
|
6-12 мес |
1,6 |
1,21 |
1,6 |
2,6-2,9 (г/кг/сутки) |
|
1-2 года |
- |
- |
- |
36 г/сут* |
|
2-3 года |
- |
- |
- |
42 г/сут |
|
1-3 года |
1,2 |
0,97 |
1,1 |
- |
|
4-6 лет |
1,1 |
0,84 |
1,0 |
54 г/сут |
|
7-10 лет |
1,0 |
0,8 |
1,0 |
63 г/сут |
|
Мальчики 11-14 лет |
1,0 |
0,79 |
1,0 |
75 г/сут |
|
Мальчики14-18 лет |
0,9 |
0,69 |
0,9 |
87 г/сут |
|
Девочки 11-14 лет |
1,0 |
0,76 |
0,95 |
69 г/сут |
|
Девочки 14-18 лет |
0,8 |
0,64 |
0,85 |
76 г/сут |
Таблица 2 - Среднее содержание лизина в белках различных продуктов [18,20].
|
Продукты |
Содержание лизина (% от общего белка) |
Лизин/белок (мг/г белка) |
|
Рыба |
9 |
90 |
|
Мясо и мясные продукты |
8 |
80 |
|
Грудное молоко |
8 |
80 |
|
Коровье молоко и молочные продукты |
7 |
70 |
|
Яйца |
6 |
60 |
|
Картофель |
6 |
60 |
|
Соя и соевые продукты |
6 |
60 |
|
Орехи |
2-8,5 |
20-85 |
|
Фрукты |
2-6,5 |
40-65 |
|
Овощи |
4-6,5 |
20-65 |
|
Хлеб и зерновые продукты |
2-4 |
20-40 |
Приложение Г3. Комплекс обязательных мероприятий, которые должны быть проведены пациенту после установления диагноза ГА1
|
Возраст больных детей |
|||||
|
|
0-6 месяцев |
7-12 месяцев |
1-3 года |
4-6 лет |
Старше 6 лет |
|
Диетотерапия |
|||||
|
Лизин белка натуральных продуктов мг/кг/сут |
100 |
90 |
80-60 |
60-50 |
Избегать чрезмерного поступления белка. Употреблять продукты с низким содержанием лизина (Dewey et al 1996) |
|
Триптофан белка натуральных продуктов мг/кг/сут |
20 |
17 |
17-13 |
13 |
13 |
|
Белок натуральный (г/кг/сут) |
1,4-1,3 |
1,5-1,3 |
1,4-1,3 |
1,3-1.1 |
1,3-1,1 |
|
Белок за счет СП* (г/кг/сут) |
1,3-0,8 |
1,0-0,8 |
0,8 |
0,8 |
0,8 |
|
Белок (общий) (г/кг/сут) |
2,7-2,1 |
2,5-2,1 |
2,2-2,1 |
2,1-1,9 |
2,1-1,9 |
|
Энергетическая ценность (ккал/кг/сут) |
115-80 |
95-80 |
95-80 |
90-78 |
90-78 |
|
Микронутриенты (%) |
?100 |
?100 |
?100 |
?100 |
>100 |
|
Основная метаболическая терапия |
|||||
|
Карнитин мг/кг/сут |
100 |
100 |
100 |
50-100 |
30-50 |
* специализированный продукт на основе аминокислот без лизина и триптофана.
Приложение Г4. Расшифровка примечаний
…ж – лекарственный препарат, входящий в Перечень жизненно необходимых и важнейших лекарственных препаратов для медицинского применения на 2016 год (Распоряжение Правительства РФ от 26.12.2015 N 2724-р)
…вк – лекарственный препарат, входящий в Перечень лекарственных препаратов для медицинского применения, в том числе лекарственных препаратов для медицинского применения, назначаемых по решению врачебных комиссий медицинских организаций (Распоряжение Правительства РФ от 26.12.2015 N 2724-р)
43
Комментарии
ПРАКТИКА ПЕДИАТРА



