Дефицит ацил-КоА дегидрогеназы жирных кислот с очень длинной углеродной цепью у детей. Клинические рекомендации.
Статьи
Дефицит ацил-КоА дегидрогеназы жирных кислот с очень длинной углеродной цепью у детей
- Союз педиатров России
Оглавление
- Ключевые слова
- Список сокращений
- Термины и определения
- 1. Краткая информация
- 2. Диагностика
- 3. Лечение
- 4. Реабилитация
- 5. Профилактика и диспансерное наблюдение
- 6. Дополнительная информация, влияющая на течение и исход заболевания
- Критерии оценки качества медицинской помощи
- Список литературы
- Приложение А1. Состав рабочей группы
- Приложение А2. Методология разработки клинических рекомендаций
- Приложение А3. Связанные документы
- Приложение Б. Алгоритмы ведения пациента
- Приложение В. Информация для пациентов
- Приложение Г.
Ключевые слова
-
Ген ACADVL
-
Ацил-КоА дегидрогеназа жирных кислот с очень длинной углеродной цепью
-
Метаболический ацидоз
-
Гипераммониемия
-
Вторичная карнитиновая недостаточность
-
Гипокетотическая гипергликемия
-
Свободный карнитин
-
Тетрадеценоилкарнитин
-
Тетрадеканоилкарнитин
-
Карнитин
-
Глицин
Список сокращений
VLCAD –very long-chain acyl-CоA dehydrogenase (VLCAD) deficiency
(дефицит ацил СоА дегидрогеназы жирных кислот с очень длинной цепью)
ДНК – дезоксирибонуклеиновая кислота
МС/МС - тандемная масс-спектрометрия
МСТ – среднецепочечные триглицериды
ТНАМ - трисамин
УЗИ – ультразвуковое исследование
ЦНС – центральная нервная система
ЭКГ - электрокардиография
ЭхоКГ – эхокардиография
Термины и определения
Метаболический криз – критическое, угрожающее жизни состояние, спровоцированное неблагоприятными факторами, обуславливающими усиление процессов клеточного катаболизма с истощением запаса углеводов и активацией метаболизма липидов, интенсификацией ?-окисления жирных кислот как резервного источника образования макроэргических соединений и проявляющееся в виде остро возникшей энцефалопатии (вялость, сонливость, летаргия, кома) и приступов рвоты. Могут наблюдаться тонико-клонические судороги, признаки сердечной и печеночной недостаточности.
Пренатальная диагностика - комплексная дородовая диагностика с целью выявления дефицита ацил-КоА дегидрогеназы жирных кислот с очень длинной углеродной цепью на стадии внутриутробного развития путем молекулярно-генетического исследования биоптата хориона с выявлением мутации гена ACADV.
1. Краткая информация
1.1 Определение
Дефицит ацил-КоА дегидрогеназы жирных кислот с очень длинной углеродной цепью – наследственное заболевание из группы дефектов митохондриального ?–окисления жирных кислот, обусловленное дефицитом указанного фермента.
1.2 Этиология и патогенез
Заболевание обусловлено мутацией гена ACADVL, который кодирует ацил-КоА дегидрогеназу жирных кислот с очень длинной углеродной цепью. Данный фермент участвует в митохондриальном ?–окислении жирных кислот, углеродная цепь которых содержит 14 – 20 атомов. Локализация гена ACADVL - 17р13. Заболевание наследуется аутосомно-рецессивно. Дефицит ацил-КоА дегидрогеназы жирных кислот с очень длинной углеродной цепью ведет к блокированию (или резкому снижению активности) митохондриального ?–окисления на уровне жирных кислот, углеродная цепь которых содержит 14–20 атомов углерода.
Это обусловливает резкое снижение кетогенеза, накопление жирных кислот с очень длинной цепью, активацию процессов ?-окисления с повышенным образованием дикарбоновых кислот. Накопление данных соединений оказывает неблагоприятный эффект на ткани головного мозга, сердца, печени, ингибирует ряд ферментов, в частности цикла синтеза мочевины и глюконеогенеза. Запуск указанных негативных процессов происходит в условиях метаболического стресса - при интеркуррентных инфекционных заболеваниях, голодании или приеме жирной пищи, физической или эмоциональной перегрузке и др. Это связано с тем, что в подобных обстоятельствах при истощении углеводных запасов необходимым источником восполнения энергетических потребностей организма становятся липиды. Однако при наличии генетически детерминированного энзимного дефекта активация катаболизма жирных кислот стимулирует образование токсичных метаболитов. Это ведет к метаболическому ацидозу, гипераммониемии, усугублению гипогликемии, поражению внутренних органов.
Запасы эндогенного карнитина тратятся на связывание токсичных производных жирных кислот, образуются ацилкарнитины. В результате в биологических жидкостях и тканях резко снижается количество свободного карнитина, развивается вторичная карнитиновая недостаточность (схема ?–окисления на жирных кислот представлена в приложении Г2) [1].
1.3 Эпидемиология
Заболевание встречается в различных популяциях. Частота среди новорожденных в странах Европы и США составляет 1:30000 - 1:50000 [10,12,13]. В Российской Федерации частота заболевания не определена.
1.4 Кодирование по МКБ-10
Е71.3 - Нарушения обмена жирных кислот.
1.5 Классификация
Выделяют следующие клинические формы болезни:
-
системное заболевание с поражением сердца и печени;
-
печеночная форма;
-
миопатическая форма.
По срокам появления первых признаков различают формы:
-
неонатальную (около ? больных);
-
детскую с манифестацией на протяжении первых двух лет жизни (около 40% больных);
-
позднюю.
2. Диагностика
2.1 Жалобы и анамнез
Ухудшение общего состояния, вялость или повышенная возбудимость, отказ от пищи, возможны судороги, ацетонемическая рвота, мышечная слабость. У детей старше 1 года – задержка психомоторного развития, умственная отсталость, судорожный синдром, нарушения функции почек, желудочно-кишечного тракта, сердечнососудистой системы, зрительного аппарата, непереносимость физических нагрузок.
В анамнезе возможны указания на родственный брак, наличие сибсов с аналогичными клиническими признаками заболевания, случаи внезапной детской смерти.
Острая метаболическая декомпенсация у детей с дефицитом ацил-КоА дегидрогеназы жирных кислот с очень длинной углеродной цепью, ведущая к критическим, угрожаемым жизни состояниям, проявляется в виде остро возникающей энцефалопатии (вялость, сонливость, летаргия, кома) и приступов рвоты. Могут наблюдаться тонико-клонические судороги, присоединяются признаки сердечной и печеночной недостаточности. Кризы обычно провоцируются неблагоприятными факторами, которые обусловливают усиление процессов клеточного катаболизма с истощением запаса углеводов и активацией метаболизма липидов, интенсификацией ?-окисления жирных кислот как резервного источника образования макроэргических соединений.
При наличии дефекта фермента, участвующего в обмене жирных кислот, такая ситуация ведет к накоплению токсичных метаболитов. Основные провоцирующие факторы:
- длительный промежуток между приемами пищи, голодание, низкая калорийность рациона;
- повышенное потребление жирной пищи;
- интеркуррентные респираторные или желудочно-кишечные инфекционные заболевания;
- физическая или психоэмоциональная нагрузка.
2.2 Физикальное обследование
Системная форма болезни характеризуется дебютом в периоде новорожденности или в раннем детском возрасте. Заболевание имеет тяжелое течение, высокую летальность (около 30%), риск внезапной детской смерти. На фоне гипогликемии и метаболического ацидоза появляется резкая мышечная гипотония, приступы рвоты и судорог, прогрессирующая вялость, сонливость. Из внутренних органов преимущественно страдают сердце и печень. Развивается гипертрофическая или дилатационная кардиомиопатия, желудочковая тахикардия, блокада сердца, гепатомегалия, жировая инфильтрация печени.
Печеночная форма также отличается ранней манифестацией, но имеет менее тяжелое течение с периодическими приступами гипокетотической гипогликемии. Миопатическая форма манифестирует у школьников или у взрослых. Ее основные проявления: непереносимость физической нагрузки, боли в мышцах, рабдомиолиз, изменение цвета мочи вследствие миоглобинурии [9,15,16,17].
Предвестниками метаболического криза, прежде всего, служат клинические признаки нарушения состояния ребенка – снижение аппетита, вялость, гипертермия. Затем появляется рвота, избыточная сонливость, может наблюдаться нарушение стула. Нарастают неврологические расстройства (вплоть до ступора или комы), появляются дыхательные и сердечно-сосудистые нарушения. Тяжесть состояния, главным образом, определяется гипогликемией (иногда может быть не резко выраженной), метаболическим ацидозом с гипокетонемией (или небольшим кетозом), гипераммониемией, обезвоживанием. Отмечено, что в раннем периоде криза данные показатели остаются нормальными за исключением равновесия кислот-оснований крови, демонстрирующего умеренный ацидоз с дефицитом оснований.
2.3 Лабораторная диагностика
Диагностика дефицита ацил-КоА дегидрогеназы жирных кислот с очень длинной углеродной цепью основана на анализе родословной, оценке данных анамнеза, клинических проявлений, результатах анализа содержания в крови тетрадеценоилкарнитина и тетрадеканоилкарнитина (С14:1 и С14), свободного карнитина (С0), определения активности фермента в культуре фибробластов и молекулярной диагностики.
-
В качестве основного метода подтверждения диагноза рекомендуется биохимический метод - тандемная масс-спектрометрия (МС/МС) [6,7,9, 12,13].
(Сила рекомендации В; Уровень доказательств II)
Комментарии: Специфические диагностические тесты это увеличение концентрации тетрадеценоилкарнитина (С14:1 – основной диагностический маркер), который обычно превышает 0,7 мкмоль/л (норма до 0,43); отмечается повышение уровня тетрадеканоилкарнитина (С14 – дополнительный диагностический маркер), иногда повышен уровень С12 и С16 - ацилкарнитинов. Кроме того, характерным лабораторным признаком является низкий показатель свободного карнитина (С0), в норме превышающий 20 мкмоль/л.
-
При изолированном выявлении низкого показателя свободного карнитина (все остальные показатели в норме) рекомендуется исследовать содержание ацилкарнитинов крови повторно после приема препаратов левокарнитина в средней суточной дозе 50 мг/кг в течение не менее 7-10 дней [6,7,8,11,13].
(Сила рекомендации В; Уровень доказательств II)
Комментарии: В ряде случаев низкий уровень свободного карнитина служит первым диагностически значимым биохимическим признаком заболевания, который указывает на выраженную недостаточность карнитина. В такой ситуации происходит тотальное снижение в биологических жидкостях концентрации всех ацилкарнитинов. Это не позволяет при тандемной масс-спектрометрии выявить значимые нарушения в спектре ацилкарнитинов и может привести к ложно-отрицательным результатам.
-
Рекомендуется определение содержания дикарбоксильных кислот с длинной углеродной цепью в моче методом газовой хроматографии масс-спектрометрии [6,13].
(Сила рекомендации С; Уровень доказательств II)
Комментарии: В моче у больных указанный показатель повышен по сравнению с референсными значениями.
-
Рекомендуется определение активности ацил-КоА дегидрогеназы жирных кислот с очень длинной углеродной цепью в фибробластах кожи [12,13 ].
(Сила рекомендации В; Уровень доказательств II)
Комментарии: исследование проводится при наличии возможностей в лаборатории, не является обязательным.
-
Для подтверждения диагноза и медико-генетического консультирования рекомендуется молекулярное исследование гена ACADVL[ 9 ].
(Сила рекомендации В; Уровень доказательств II)
Комментарии: Обследованию на дефицит ацил-КоА дегидрогеназы жирных кислот с очень длинной углеродной цепью подлежат следующие группы детей:
-
дети любого возраста из семей, имеющих больных с данным заболеванием (в первую очередь, братья и сестры больного) или имеющих случаи внезапной детской смерти;
-
дети первых дней/недель и месяцев жизни с гипогликемией, ацидозом, приступами рвоты и судорог, поражением сердца и печени;
-
дети любого возраста с повторными приступами рвоты, гипотонии, поражением сердца и печени;
-
дети старшего возраста и взрослые с непереносимостью физической нагрузки, приступами боли в мышцах, рабдомиолизом, миоглобинурией.
-
Рекомендуется в межприступный период в процессе комплексного лечения контролировать лабораторные показатели не реже 1 раза в год, параметры кислотно-основного состояния – не реже 1 раза в 6 месяцев [12].
(Сила рекомендации С; Уровень доказательств II)
Комментарии: Проводят контроль клинического анализа крови, уровня гемоглобина, общего белка, альбумина, глюкозы, билирубина, трансаминаз, креатинфосфокиназы, сывороточного железа, эссенциальных жирных кислот, свободного карнитина и ацилкарнитинов, параметры кислотно-основного состояния крови, определение содержания органических кислот в моче.
Неблагоприятные лабораторные признаки, свидетельствующие о несбалансированности терапии и угрозе развития метаболического криза: уменьшение содержания свободного карнитина, глюкозы, тенденция к снижению pH крови, дефициту оснований, нарастание содержания ацилкарнитинов с очень длинной цепью (при снижении показателя свободного карнитина) в крови и повышение экскреции органических кислот.
На недостаточность нутритивной поддержки указывает снижение уровня гемоглобина, железа, общего белка и альбумина. Низкий уровень в крови эссенциальных жирных кислот (линолевой, линоленовой, арахидоновой, докозагексаеновой и др.), нарушенное соотношение линолевой/линоленовой кислот (в норме 5:1 – 10:1) могут обусловить задержку роста, дистрофические изменения кожи и волос.
2.4 Инструментальная диагностика
-
Рекомендуются рентгенография грудной клетки, электрокардиография (ЭКГ), эхокардиография (ЭхоКГ) для исключения или подтверждения) патологии сердца [3,12,13, 15].
(Сила рекомендации С; Уровень доказательств II)
-
Рекомендуется ультразвуковое исследование (УЗИ) органов брюшной полости и почек для исключения (или подтверждения) патологии печени и почек [12,13, 15].
(Сила рекомендации С; Уровень доказательств II)
-
Рекомендуется проведение офтальмоскопии при подозрении на патологию зрительного анализатора [12,13, 15].
(Сила рекомендации С; Уровень доказательств II)
2.5 Иная диагностика
-
Рекомендуется консультативная помощь специалистов офтальмолога, кардиолога, гастроентеролога, генетика, диетолога, невропатолога [12,13, 15].
(Сила рекомендации С; Уровень доказательств II)
Комментарии: По показаниям проводятся консультации гематолога, ортопеда, психолога.
2.6. Дифференциальная диагностика
-
Рекомендуется проводить дифференциальную диагностику с гипоксическими поражениями нервной системы, внутриутробными инфекциями, поствакцинальными осложнениями, наследственными нарушениями обмена веществ, в частности органическими ацидемиями, дефектами цикла синтеза мочевины, другими формами дефектов транспорта и ?-окисления жирных кислот, а также с кардиомиопатиями, гепатитами различного происхождения [4,9,10,11 ].
(Сила рекомендации С; Уровень доказательств II)
3. Лечение
3.1 Консервативное лечение
Стратегия лечения пациентов с наследственным дефектом ацил-КоА дегидрогеназы жирных кислот с очень длинной углеродной цепью носит комплексный характер, в его основе лежит коррекция метаболических нарушений посредством диеты, которая заключается в снижении потребления больным пищевых жиров в качестве резервной составляющей тканевой биоэнергетики, минимизации катаболизма жирных кислот и уменьшении их значимости для восполнения энергозатрат клетки с обеспечением нормальных процессов анаболизма, роста и нутритивного статуса детей. Главная задача диетотерапии – это профилактика голодания, предупреждение гипогликемии и минимально допустимое снижение поступления с пищей патогенетически значимых жирных кислот (в данном случае жирных кислот с очень длинной углеродной цепью) и их источников.
-
Рекомендуется начать диетическое и симптоматическое лечение при первом подозрении на дефицит ацил-КоА дегидрогеназы жирных кислот с очень длинной углеродной цепью до получения подтверждающих результатов диагностики [5,12,13,14 ].
(Сила рекомендации С; Уровень доказательств II)
-
В зависимости от состояния ребенка питание рекомендовано осуществлять энтеральным путем, а также через зонд или гастростому, расчет проводится строго индивидуально.
(Сила рекомендации С; Уровень доказательств II)
Комментарии: Диетотерапия зависит от состояния больного (межприступный период или состояние метаболического криза).
-
В межприступный период рекомендовано соблюдать основные правила диетотерапии:
(Сила рекомендации С; Уровень доказательств II)
-
исключение из питания женского молока, детских молочных смесей, молока других животных;
-
использование специализированных безжировых смесей (Приложение Г1);
-
соблюдение режима кормлений (строго по часам): для детей грудного возраста промежутки между кормлениями - не более 3-х часов, для детей 1 года жизни – не более 4-х часов;
-
обязательные ночные кормления (с использованием мальтодекстрина или кукурузного крахмала для детей с 2-х летнего возраста из расчета 2-2,5 г/кг массы тела);
-
поддержание энергетической ценности пищевого рациона не ниже 100 ккал/кг для детей грудного и раннего возраста;
-
низкое содержание жиров (для детей грудного возраста не более 25% от энергетической ценности всего рациона, для детей 1 года жизни – не более 20%);
-
жировой компонент рациона должен быть представлен преимущественно среднецепочечными триглицеридами - от 15-18% энергетической ценности рациона на первом году жизни (около 2 г/кг массы тела) до 10-15% у детей старше 1 года (не более 1,2-1,3 г/кг массы тела), доля энергоценности рациона рассчитывается от рекомендуемой для здоровых детей [2];
-
возможно минимальное поступление эссенциальных жирных кислот (особенно ?-линоленовой) - до 3% от общей энергетической ценности рациона, чтобы избежать глубокого дефицита;
-
профилактика стрессовых состояний, способных провоцировать метаболический криз (инфекционные заболевания, хирургические вмешательства, психоэмоциональные стрессы и т.п.).
-
Рекомендовано в качестве основных пищевых источникв среднецепочечных жирных кислот использовать в питании пациентов:
-
50% эмульсия среднецепочечных триглицеридов (MCT - Medium Chain Triglycerides);
-
натуральные растительные масла (кокосовое масло, масло грецкого ореха).
(Сила рекомендации С; Уровень доказательств II)
Комментрии: Для всех пациентов независимо от возраста требуется индивидуальное составление рациона, для детей первого года жизни рекомендуется использование специализированных смесей с жировым компонентом, представленным среднецепочечными триглицеридами. Перечень продуктов, подлежащих исключению из рациона представлен в Приложении Г1
-
Рекомендуется с осторожностью назначать препараты левокарнитина [ 5,12 ].
(Сила рекомендации С; Уровень доказательств II)
Комментарии: Левокарнитин назначают с целью усиления связывания метаболитов жирных кислот и ликвидации карнитиновой недостаточности больным. Применение препаратов карнитина обосновано патогенезом имеющихся метаболических расстройств, и низкими показателями уровня свободного карнитина в крови. Однако, учитывая сведения о возможном неблагоприятном эффекте препаратов левокарнитина у детей с нарушением обмена жирных кислот с длинной цепью в связи с избыточным накоплением токсичных длинноцепочечных ацилкарнитинов, вначале рекомендуется использовать небольшие дозы препарата (до 20 мг/кг) с последующим увеличением до 50 мг/кг массы тела в сутки (в некоторых случаях до 80 мг/кг) за 2-3 приема.
-
Рекомендуется в комплексном лечении назначать глицин ж [5,12].
(Сила рекомендации С; Уровень доказательств II)
Комментарии: Глицин ж подобно карнитину, обладает способностью конъюгировать дериваты жирных кислот. Суточная доза в среднем составляет 200-600 мг в течение 2-3 месяцев.
-
Рекомендуется по показаниям назначение витаминов группы В и жирорастворимых витаминов А, D, Е [5,12].
(Сила рекомендации С; Уровень доказательств II)
Комментарии: Витамины назначают в возрастных профилактических дозах курсами по 2-3 месяца.
-
Рекомендуется по показаниям симптоматическое лечение ноотропными препараты и гепатопротекторами [5,12].
(Сила рекомендации С; Уровень доказательств II)
-
Не рекомендуется использовать препараты вальпроевой кислотыж в случае необходимости назначения антиконвульсантов [5,12].
(Сила рекомендации С; Уровень доказательств II)
Комментарии: У пациентов с дефектами митохондриального ?-окисления жирных кислот применение вальпроатов может привести к развитию тяжелой печеночной недостаточности.
3.2 Лечение в период метаболического криза
Родители и пациенты должны быть проинформированы об основных принципах лечения заболевания.
Состояние метаболического криза является показанием для госпитализации и проведения интенсивной, в том числе инфузионной терапии, которая должна начинаться незамедлительно. Тактика лечения детей в период криза включает дополнительное введение глюкозы для энергетической поддержки и уменьшения интенсивности процессов катаболизма, коррекцию метаболического ацидоза, гипераммониемии и водно-электролитных нарушений, активацию связывания накапливающихся дериватов жирных кислот путем увеличения дозы левокарнитина, коррекцию диетотерапии.
Устранение гипогликемии и энергетической недостаточности имеет первостепенное значение для сохранения жизни и здоровья детей.
-
Рекомендуется внутривенное введение раствора 10% декстрозы ж из расчета 7 – 10 мг/кг/мин под контролем ее уровня в крови [5,12].
(Сила рекомендации С; Уровень доказательств II)
Комментарии: При выраженной гипогликемии инфузию начинают с 25% раствора декстрозы (2 мл/кг), далее продолжают вводить 10% раствор декстрозы со скоростью 7-8 мг/кг/мин. Назначение раствора декстрозы не только восполняет тканевой энергетический дефицит, но и подавляет липолиз и снижает продукцию токсичных дериватов жирных кислот.
-
Рекомендуется коррекция метаболического ацидоза (при уровне бикарбонатов сыворотки крови <16 мЭкв/л) [5,12].
(Сила рекомендации С; Уровень доказательств II)
Комментарии: Дефицит бикарбонатов купируется путем внутривенного введения щелочных растворов: гидрокарбоната натрияж (код АТХ - В05ХА), Калия хлорид+Натрия гидрокарбонат+Натрия хлорид или (трисбуфера) трисамина (ТНАМ). Гидрокарбонат натрия применяется в виде 8,4% и 4,2% раствора для удобства перерасчета на ммоль NаНСО. Его дозировка (ммоль) определяется по формуле: (-ВЕ) Х масса тела (кг) Х 0,3. Также больным назначают щелочное питье – раствор соды из расчета ?-1 чайная ложка на 200 мл воды, щелочные минеральные воды.
-
Рекомендуется в зависимости от тяжести состояния каждые 6-12 часов контролировать показатели кислотно-основного состояния крови, уровня натрия и калия в крови [5,12,13,14].
(Сила рекомендации С; Уровень доказательств II)
-
Рекомендуется коррекция водно-электролитных нарушений и гипераммониемии[5,12].
(Сила рекомендации С; Уровень доказательств II)
Комментарии: При сохраняющейся гипогидратации проводится путем внутривенного введения физиологического раствора. Однако необходимо иметь в виду, что главным мероприятием в комплексе интенсивной терапии является введение растворов глюкозы и щелочных растворов.
-
Рекомендуются назначение аргинина или цитруллина при уровне аммиака в крови выше 200 мкмоль/л [5, 12].
(Сила рекомендации С; Уровень доказательств II)
Комментарии: Дополнительное назначение 250-300 мг/кг аргинина или 350 мг/кг цитруллина стимулирует синтез мочевины.
-
Вопрос о назначении или увеличении дозы левокарнитина рекомендуется решать строго индивидуально [5,12].
(Сила рекомендации С; Уровень доказательств II)
Комментарии: Левокарнитин активирует связывание накапливающихся органических кислот при увеличении суточной дозы карнитина до 80-100 мг/кг, однако дотация карнитином может привести к повышенному продуцированию ацилкарнитинов с очень длинной цепью, которые способны спровоцировать развитие сердечной аритмии.
-
Рекомендуется коррекция диетических мероприятий в период метаболического криза [14].
(Сила рекомендации С; Уровень доказательств II)
Комментарии: Диетические ограничения при кризе:
-
полностью исключить потребление жиров на период острого криза (на 24-48 час.), далее вводить минимальное количество среднецепочечных жиров при сохранении высокой калорийности (не менее 100–115 ккал/кг) рациона в основном за счет углеводов;
-
избегать голодания, когда прекращаются инфузии.
-
Рекомендуется использовать приемлемый для ребенка способ кормления (энтеральный самостоятельно, через зонд или гастростому) [14].
(Сила рекомендации С; Уровень доказательств II)
3.3 Хирургическое лечение
-
При возникновении показаний для хирургических вмешательств рекомендуется коллегиальное принятие решения о тактике его проведения [5,12].
(Сила рекомендации С; Уровень доказательств II)
4. Реабилитация
-
Рекомендуются реабилитационные мероприятия, направленные на коррекцию кардиологических, офтальмологических и неврвнопсихических нарушений [4,5,12].
(Сила рекомендации С; Уровень доказательств II)
5. Профилактика и диспансерное наблюдение
Профилактические меры включают медико-генетическое консультирование семей и пренатальную диагностику, которая проводится с помощью молекулярно-генетического исследования биоптата хориона с выявлением мутации гена
Неонатальный скрининг в Российской Федерации не проводится.
Продолжительность госпитализации зависит от скорости верификации диагноза, коррекции метаболических нарушений путем проведения интенсивной терапии и начала патогенетической диетотерапии, а также от сроков появления положительной динамики со стороны центральной нервной системы и других органов, скорости восстановления показателей глюкозы крови и кислотно-щелочного состояния, ответ на лечение отмечается в течение 5-7 дней. Пребывание в стационаре в среднем составляет 21 день.
После выписки из стационара дети нуждаются в наблюдении педиатра, невролога, кардиолога, гастроэнтеролога, офтальмолога, диетолога, генетика, проведении ЭКГ (по показаниям - Холтеровского мониторирования ЭКГ), Эхо-КГ, УЗИ органов брюшной полости не реже 2 раз в год.
Ежемесячно необходимо проводить коррекцию лечебного питания и симптоматической терапии, 1 раз в 6-12 месяцев рекомендуется контролировать содержание патологических метаболитов в сыворотке крови или моче методом тандемной масс-спектрометрии.
Ежегодно дети должны проходить углубленную диспансеризацию в условиях дневного стационара (длительность госпитализации не менее 10 суток), где также осуществляются необходимые реабилитационные мероприятия.
Родители должны быть обучены правилам организации терапии в межприступный период и в период угрозы метаболического криза. У родителей ребенка и при ребенке всегда должна быть памятка с указанием неотложных мероприятий в период начинающегося метаболического криза.
6. Дополнительная информация, влияющая на течение и исход заболевания
Прогноз состояния и уровня психического развития пациентов зависит от тяжести заболевания, степени поражения внутренних органов (сердце, печень) и ЦНС, сроков начала лечения и эффективности интенсивной терапии при метаболической декомпенсации. Рано манифестирующая системная форма заболевания обычно имеет более тяжелое течение и менее благоприятный прогноз.
Критерии оценки качества медицинской помощи
Список литературы
-
МP 2.3.1.2432-08 "Нормы физиологических потребностей в энергии и пищевых веществах для различных групп населения Российской Федерации" (утв. Главным государственным санитарным врачом РФ 18 декабря 2008 г.).
-
Sharef Waadallah Sharef, Khalfan Al-Senaidi, and Surendra Nath Joshi. Successful Treatment of Cardiomyopathy due to Very Long-Chain Acyl-CoA Dehydrogenase Deficiency: First Case Report from Oman with Literature Review. Oman Medical Journal (2013) Vol. 28, No. 5:354-356.
-
Diekman EF, Ferdinandusse S, van der Pol L, Waterham HR, Ruiter JP, Ijlst L, Wanders RJ, Houten SM, Wijburg FA, Blank AC, Asselbergs FW, Houtkooper RH, Visser G. Fatty acid oxidation flux predicts the clinical severity of VLCAD deficiency. Genet Med. 2015 Apr 2.
-
Tenopoulou M, Chen J, Bastin J, Bennett MJ, Ischiropoulos H, Doulias PT. Strategies for correcting very long chain acyl-CoA dehydrogenase deficiency. J Biol Chem. 2015 Apr 17;290(16):10486-94.
-
Top?u Y, Bayram E, Karao?lu P, Yi? U, Kurul SH. Importance of acylcarnitine profile analysis for disorders of lipid metabolism in adolescent patients with recurrent rhabdomyolysis: Report of two cases. Ann Indian Acad Neurol. 2014 Oct;17(4):437-40.
-
Berardo A, DiMauro S, Hirano M. A diagnostic algorithm for metabolic myopathies. Curr Neurol Neurosci Rep. 2010;10:118–26.
-
Kilfoyle D, Hutchinson D, Potter H, George P. Recurrent myoglobinuria due to carnitine palmitoyltransferase II deficiency: Clinical, biochemical, and genetic features of adult-onset cases. N Z Med J. 2005;118:U1320.
-
Deschauer M, Wieser T, Zierz S. Muscle carnitine palmitoyltransferase II deficiency: Clinical and molecular genetic features and diagnostic aspects. Arch Neurol. 2005;62:37–41.
-
Elsayed EF, Reilly RF. Rhabdomyolysis: A review, with emphasis on the pediatric population. Pediatr Nephrol.2010;25:7–18.
-
Moore SJ, Haites NE, Broom I, White I, Coleman RJ, Pourfarzam M, et al. Acylcarnitine analysis in the investigation of myopathy. J Inherit Metab Dis. 1998;21:427–8.
-
Solis JO, Singh RH. Management of fatty acid oxidation disorders: A survey of current treatment strategies. J Am Diet Assoc. 2002;102:1800–3.
-
Al-Thihli K, Sinclair G, Sirrs S, Mezei M, Nelson J, Vallance H. Performance of serum and dried blood spot acylcarnitine profiles for detection of fatty acid ?-oxidation disorders in adult patients with rhabdomyolysis. J Inherit Metab Dis. 2014. Mar;37(2):207-13.
-
Diekman EF, van Weeghel M, Wanders RJ, Visser G, Houten SM. Food withdrawal lowers energy expenditure and induces inactivity in long-chain fatty acid oxidation-deficient mouse models. FASEB J. 2014 Jul;28(7):2891-900.
-
Oliveira SF, Pinho L, Rocha H, Nogueira C, Vilarinho L, Dinis MJ, Silva C. Rhabdomyolysis as a presenting manifestation of very long-chain acyl-coenzyme a dehydrogenase deficiency. Clin Pract. 2013 Aug 6;3(2):e22.
-
Tucci S, Fl?gel U, Hermann S, Sturm M, Sch?fers M, Spiekerkoetter U. Development and pathomechanisms of cardiomyopathy in very long-chain acyl-CoA dehydrogenase deficient (VLCAD(-/-)) mice. Biochim Biophys Acta. 2014 May;1842(5):677-85.
-
Xiong D, He H, James J, Tokunaga C, Powers C, Huang Y, Osinska H, Towbin JA, Purevjav E, Balschi JA, Javadov S, McGowan FX Jr, Strauss AW, Khuchua Z. Cardiac-specific VLCAD deficiency induces dilated cardiomyopathy and cold intolerance. Am J Physiol Heart Circ Physiol. 2014 Feb;306(3):H326-38.
-
Биохимия: Учебник для вузов под ред. Е.С. Северина., 2003. 779 (с. http://biochemistry.ru/biohimija_severina/B5873Part62-399.html).
Приложение А1. Состав рабочей группы
-
Баранов А.А. – академик РАН, профессор, д.м.н., Председатель Исполкома Союза педиатров России.
-
Намазова-Баранова Л.С. - член-корр. РАН, проф., д.м.н. заместитель Председателя Исполкома Союза педиатров России.
-
Боровик Т.Э. – д.м.н., проф., член Исполкома Союза педиатров России.
-
Бушуева Т.В. – д.м.н., член Союза педиатров России.
-
Глоба О.В. - к.м.н., член Союза педиатров России.
-
Журкова Н.В. – к.м.н., член Союза педиатров России.
-
Захарова Е.Ю. – д.м.н., проф., член Ассоциации медицинских генетиков.
-
Звонкова Н.Г. - к.м.н., член Союза педиатров России.
-
Кузенкова Л.М. - д.м.н., проф., член Исполкома Союза педиатров России.
-
Куцев С.И. - д.м.н., проф., член-корр. РАН, президент Ассоциации медицинских генетиков.
-
Николаева Е.А. - д.м.н., проф., член Ассоциации медицинских генетиков.
-
Новиков П.В. - д.м.н., проф., член Ассоциации медицинских генетиков.
-
Пушков А.А. - к.б.н., член Союза педиатров России.
-
Савостьянов К.В. - к.б.н., член Союза педиатров России.
Авторы подтверждают отсутствие финансовой поддержки/конфликта интересов, который необходимо обнародовать.
Приложение А2. Методология разработки клинических рекомендаций
Целевая аудитория данных клинических рекомендаций:
-
педиатры
-
врачи общей семейной практики (семейная медицина)
-
генетики
-
диетологи
-
неврологи
-
медицинские психологи
-
дефектологи
-
студенты медицинских ВУЗов, интерны, ординаторы;
Таблица П1 – Уровни достоверности доказательств
|
Уровень достоверности |
Источник доказательств |
|
I (1) |
Проспективные рандомизированные контролируемые исследования Достаточное количество исследований с достаточной мощностью, с участием большого количества пациентов и получением большого количества данных Крупные мета-анализы Как минимум одно хорошо организованное рандомизированное контролируемое исследование Репрезентативная выборка пациентов |
|
II (2) |
Проспективные с рандомизацией или без исследования с ограниченным количеством данных Несколько исследований с небольшим количеством пациентов Хорошо организованное проспективное исследование когорты Мета-анализы ограничены, но проведены на хорошем уровне Результаты не презентативны в отношении целевой популяции Хорошо организованные исследования «случай-контроль» |
|
III (3) |
Нерандомизированные контролируемые исследования Исследования с недостаточным контролем Рандомизированные клинические исследования с как минимум 1 значительной или как минимум 3 незначительными методологическими ошибками Ретроспективные или наблюдательные исследования Серия клинических наблюдений Противоречивые данные, не позволяющие сформировать окончательную рекомендацию |
|
IV (4) |
Мнение эксперта/данные из отчета экспертной комиссии, экспериментально подтвержденные и теоретически обоснованные |
Таблица П2 – Сила рекомендаций
|
Уровень убедительности |
Описание |
Расшифровка |
|
A |
Рекомендация основана на высоком уровне доказательности (как минимум 1 убедительная публикация I уровня доказательности, показывающая значительное превосходство пользы над риском) |
Метод/терапия первой линии; либо в сочетании со стандартной методикой/терапией |
|
B |
Рекомендация основана на среднем уровне доказательности (как минимум 1 убедительная публикация II уровня доказательности, показывающая значительное превосходство пользы над риском) |
Метод/терапия второй линии; либо при отказе, противопоказании, или неэффективности стандартной методики/терапии. Рекомендуется мониторирование побочных явлений |
|
C |
Рекомендация основана на слабом уровне доказательности (но как минимум 1 убедительная публикация III уровня доказательности, показывающая значительное превосходство пользы над риском) или нет убедительных данных ни о пользе, ни о риске) |
Нет возражений против данного метода/терапии или нет возражений против продолжения данного метода/терапии Рекомендовано при отказе, противопоказании, или неэффективности стандартной методики/терапии, при условии отсутствия побочных эффектов |
|
D |
Отсутствие убедительных публикаций I, II или III уровня доказательности, показывающих значительное превосходство пользы над риском, либо убедительные публикации I, II или III уровня доказательности, показывающие значительное превосходство риска над пользой |
Не рекомендовано |
Актуализация данных клинических рекомендаций будет проводиться не реже, чем один раз в три года. Принятие решения об обновлении будет принято на основании предложений, представленных медицинскими профессиональными некоммерческими организациями с учётом результатов комплексной оценки лекарственных препаратов, медицинских изделий, а также результатов клинической апробации.
Приложение А3. Связанные документы
-
Приказ Минздравсоцразвития РФ № 185 от 22.03.2006 года «О массовом обследовании новорожденных детей на наследственные заболевания»,
-
Приказ Министерства здравоохранения и социального развития РФ от 16 апреля 2012 г. N 366н "Об утверждении Порядка оказания педиатрической помощи"
-
Приказ Министерства здравоохранения РФ "Об утверждении Порядка оказания медицинской помощи больным с врожденными и (или) наследственными заболеваниями" от 15 ноября 2012 г. N 917н
-
Постановление Правительства Российской Федерации от 9 апреля 2015 года №333 "Об утверждении Правил формирования перечня специализированных продуктов лечебного питания для детей-инвалидов"
Приложение Б. Алгоритмы ведения пациента
Пациент с подозрением
на дефицит ацил КоА дегидрогеназы жирных кислот с очень длинной цепью
Диагностика

Консультация профильного специалиста
НЕТ
Терапия в стационаре
Диспансерное наблюдение и лечение с периодическим контролем в специализированном педиатрическом отделении


ДА НЕТ 

Приложение В. Информация для пациентов
Нарушение ?-окисления жирных кислот с очень длинной цепью или дефицит ацил-КоА дегидрогеназы жирных кислот с очень длинной углеродной цепью относится к группе наследственных митохондриальных дефектов.
Дети с подозрением на дефицит ацил-КоА дегидрогеназы ЖКОДЦ требуют тщательного обследования и наблюдения. Семьи, в которых есть больные с подобным установленным диагнозом ПА, должны пройти медико-генетическое консультирование.
В клинической картине преобладают задержка психомоторного развития, поражение сердца (кардиомиопатии), печени (печеночная форма) или смешанная форма с поражением нескольких органов.
Лечение – низкожировая диета с исключением продуктов, богатых жирными кислотами с очень длинной цепью, их перечень представлен в приложении Г1.
Важно, что из питания детей первого года жизни полностью исключаются материнское молоко и детские адаптированные смеси, так как они являются источником длинноцепочечных жирных кислот. В этих случаях используются специализированные лечебные смеси, не содержащие жира, а в качестве жировой добавки используются только среднецепочечные жиры, продукты их содержащие или специальные жировые эмульсии (приложение Г1).
Наиболее опасными для жизни состояниями, ведущими при отсутствии лечения к необратимым последствиям вплоть до летального исхода, являются метаболические кризы, соповождающиеся снижением уровня сахара в крови (гипогликемия).
Кризы обычно провоцируются такими неблагоприятными факторами, как нарушение диеты, пренебрежение назначениями врача, вирусные и бактериальные инфекции, стрессовые ситуации, травмы и, хирургические вмешательства, эмоциональные и физически нагрузки.
Предвестниками криза является снижение эмоционального тонуса, вялость, сонливость, далее ребенок отказывается от еды, может быть рвота, возможен подъем температуры, особенно при дебюте инфекционного заболевания.
При первых симптомах метаболического криза необходимо срочно госпитализировать ребенка, до приезда врача скорой помощи незамедлительно начать терапию на дому.
Терапия на дому
При наличии фебрильной лихорадки ниже 38,50С и отсутствии таких симптомов как, рвота, отказ от еды и различных неврологических нарушений показано продолжение основной метаболической терапии и максимальное ограничение поступления натурального белка с пищей на срок до 12 часов, госпитализация больного.
При наличии температуры выше 38,50С дают жаропонижающие средства, в том числе, ибупрофен, в дозе 10-15 мг/кг/сутки, максимальное назначение препарата до 3-4 раз в сутки и не более чем 60 мг/сутки, обеспечивают достаточное поступление жидкости, при этом используют раствор глюкозы и мальтодекстрин (при их отсутствии - сладкий компот, кисель) в объемах, указанных в таблице 13.
При стабильном состоянии пациента на протяжении всего наблюдаемого периода продолжают плановую симптоматическую терапию.
Важно с осторожностью отнестись к использованию L карнитина С появлением первых признаков метаболического криза, не дожидаясь прихода врача, следует увеличить дозу перорального через рот) до 200 мг/мг/сутки.
Таблица 1 - Основные принципы терапии на дому
|
А. Углеводы |
Мальтодекстрин |
|
Возраст |
Объем жидкости (мл) в день через рот |
|
0-1 |
Минимально. 150 мл/кг |
|
1-2 |
120 мл/кг |
|
2-6 |
1200-1500 в сутки |
|
Старше 6 |
Интенсивная терапия продолжается в том же объеме, что и в возрасте 0-6 лет, возможна индивидуальная адаптация объема и дозы. |
Приложение Г.
Приложение Г1. Основные пищевые источники жирных кислот с очень длинной цепью
Жирные кислоты с очень длинной цепью содержатся во многих продуктах детского питания, ежедневно используются в рационе. Однако при нарушении окисления жирных кислот они должны быть полностью исключены из пищи ребенка. К таким продуктам относятся
-
женское молоко;
-
стандартные детские молочные смеси;
-
рыба;
-
морепродукты;
-
бурые морские водоросли;
-
рыбий жир;
-
специальные пищевые добавки, содержащие рыбий жир или длинноцепочечные полиненасыщенные жирные кислоты;
-
растительные жиры, богатые жирными кислотами с очень длинной цепью (низкоолеиновое подсолнечное, рапсовое, кукурузное масла).
-
некоторые продукты животного происхождения - молоко, творожные изделия, кисломолочные продукты, мясо свинины, сало, колбасы;
Для вскармливания детей первого года жизни применяют специализированные смеси, в которых жиры не содержатся или представлены в незначительном количестве (табл.1). При этом по остальным основным и минорным пищевым компонентам смеси удовлетворяют потребности грудных детей. Для компенсации жирового компонента используются жировые эмульсии (специализированные продукты) или натуральные масла, являющиеся источником среднецепочечных (СЦТ) жирных кислот (кокосовое и пальмовое масла).
Таблица 1- Информация о продуктах
|
продукт |
Содержание на 100 г сухого продукта |
|||
|
Белки,г |
Жиры,г |
Углеводы,г |
ккал |
|
|
Продукты с минимальным содержанием жира |
||||
|
Моноген* |
11,4 |
11, в т.ч. 20%- длинноцепочечные 80%-среднецепочечные |
68 |
424 |
|
Basic F** |
14 |
Менее 0,5,в т.ч. линолевая?0,04 ?-линоленовая ?0,004 |
79 |
374 |
|
Жировая эмульсия среднецепочечных жирных кислот |
||||
|
Ликвиджен*** |
- |
50,0, В.т.ч.49,2 СЦТ |
- |
450 на 100 мл |
* регистрация продукта в РФ планируется в 2017 году, зарегистрирован в странах ЕС.
** продукт зарегистрирован только в странах ЕС, регистрация в РФ не планируется.
***продукт включен в Перечень специализированных продуктов для детей инвалидов.
Приложение Г2. Последовательность реакций ?-окисления жирных кислот*
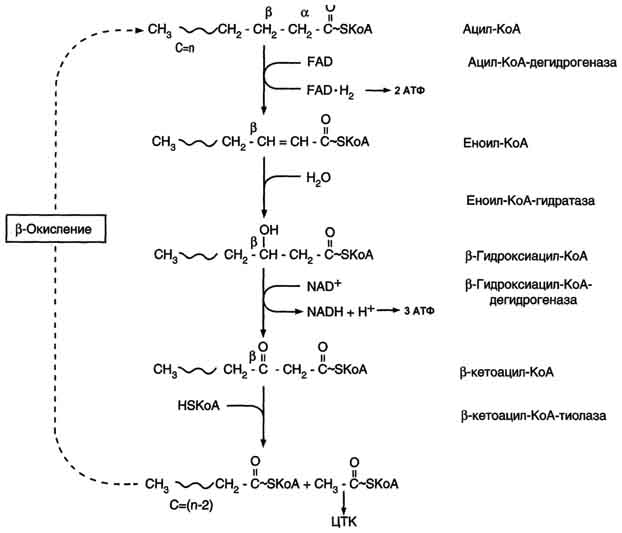
* одни и те же реакции повторяются с радикалом жирной кислоты до тех пор, пока вся кислота не превратится в ацетильные остатки.
Приложение Г3. Расшифровка примечаний
…ж – лекарственный препарат, входящий в Перечень жизненно необходимых и важнейших лекарственных препаратов для медицинского применения на 2016 год (Распоряжение Правительства РФ от 26.12.2015 N 2724-р)
…вк – лекарственный препарат, входящий в Перечень лекарственных препаратов для медицинского применения, в том числе лекарственных препаратов для медицинского применения, назначаемых по решению врачебных комиссий медицинских организаций (Распоряжение Правительства РФ от 26.12.2015 N 2724-р)
33
Комментарии
ПРАКТИКА ПЕДИАТРА



