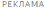

Эврисди - инструкция по применению
См. откуда получены инструкции МЕДИ РУ
Регистрационный номер
ЛП-006602
Торговое наименование
Эврисди®
Международное непатентованное или группировочное наименование
Рисдиплам
Лекарственная форма
Порошок для приготовления раствора для приема внутрь
Состав
Один флакон (2 г порошка для приготовления раствора для приема внутрь) содержит:
действующее вещество: рисдиплам – 60 мг;
вспомогательные вещества: маннитол – 1344.70 мг, изомальт – 237.25 мг, ароматизатор клубничный (мальтодекстрин кукурузный, крахмал кукурузный восковой модифицированный (Е1450), ароматизирующие компоненты) – 150.00 мг, винная кислота – 120.50 мг, натрия бензоат – 30.00 мг, макрогол/полиэтиленгликоль 6000 – 20.00 мг, сукралоза – 16.00 мг, аскорбиновая кислота – 14.10 мг, динатрия эдетата дигидрат – 7.45 мг.
1 мл приготовленного (восстановленного) раствора содержит 0.75 мг рисдиплама.
Описание
Порошок или порошок с комками от светло-желтого до желтого с сероватым или зеленоватым оттенком или светло-зеленого с желтоватым оттенком цвета (при выпуске). Порошок или порошок с комками, или скомковавшийся порошок от светло-желтого до желтого с сероватым или зеленоватым оттенком или светло-зеленого с желтоватым оттенком цвета (в течение срока годности).
Приготовленный (восстановленный) раствор
Прозрачный раствор от зеленовато-желтого до желтого цвета.
Фармакотерапевтическая группа
Прочие препараты для лечения заболеваний костно-мышечной системы.
Код АТХ
М09АХ10
Фармакологические свойства
Фармакодинамика
Механизм действия
Рисдиплам представляет собой модификатор сплайсинга предшественника матричной рибонуклеиновой кислоты (пре-мРНК) гена выживаемости двигательных нейронов 2 (SMN2), разработанный для лечения спинальной мышечной атрофии (СМА), причиной которой являются мутации в хромосоме 5q, что приводит к недостаточности белка SMN. Недостаточность функционального белка SMN является патофизиологическим механизмом развития СМА всех типов. Рисдиплам корректирует сплайсинг SMN2, сдвигая баланс с исключения экзона 7 на включение экзона 7 в транскрипте м-РНК. приводя к образованию функционального и стабильного белка SMN. Таким образом, рисдиплам лечит СМА путем увеличения и сохранения уровней функционального белка SMN.
Рисдиплам равномерно распределяется во всем организме, в том числе в центральной нервной системе (ЦНС), проникая через гематоэнцефалический барьер и соответственно приводя к увеличению уровня белка SMN в ЦНС и по всему организму. Концентрации рисдиплама в плазме и белка SMN в крови отражают распределение и фармакодинамические эффекты рисдиплама в тканях, а именно: мышечных тканях и тканях мозга.
Во всех клинических исследованиях применение рисдиплама приводило к устойчивому и длительному увеличению уровня белка SMN. В течение 4 недель после начала лечения медианный уровень белка SMN был более чем в 2 раза выше по сравнению с исходным значением, согласно измерениям в крови. Повышение уровня белка SMN сохранялось на протяжении периода лечения ≤2 лет у пациентов с манифестацией СМА в младенческом возрасте и у пациентов с поздней манифестацией СМА (см. подраздел «Клиническая эффективность»).
Клиническая эффективность
Эффективность препарата Эврисди® для лечения пациентов с манифестацией СМА в младенческом возрасте и пациентов с поздней манифестацией СМА оценивалось в 2 опорных клинических исследованиях FIREFISH и SUNFISH и подтверждено дополнительными данными, полученными в исследовании JEWELFISH. В целом результаты исследований подтверждают эффективность препарата Эврисди® у пациентов со СМА.
Манифестация СМА в младенческом возрасте
ВР39056 (FIREFISH) представляет собой открытое исследование по изучению эффективности, безопасности, фармакокинетики и фармакодинамики препарата Эврисди® у пациентов с симптоматической СМА 1 типа (у всех пациентов было генетически подтверждено заболевание с двумя копиями гена SMN2). Исследование состоит из двух частей. Часть 1 исследования FIREFISH разработана как часть исследования по подбору дозы. В подтверждающей части 2 исследования FIREFISH оценивалась эффективность препарата Эврисди® в терапевтической дозе, которая была выбрана на основании результатов части 1 исследования (см. раздел «Способ применения и дозы»). Пациенты из части 1 не принимали участия в части 2.
В частях 1 и 2 ключевой конечной точкой по эффективности была возможность сидеть без поддержки в течение, как минимум, 5 секунд согласно измерениям по пункту 22 шкалы развития младенцев и детей Бейли – 3 издание (Bayley Scales of Infant and Toddler Development, BSID-III, шкала крупной моторики) после 12 месяцев лечения препаратом Эврисди®.
FIREFISH часть 2
В часть 2 исследования FIREFISH был набран 41 пациент со СМА 1 типа.
Медиана возраста возникновения клинических признаков и симптомов СМА 1 типа составила 1.5 месяцев (1.0-3.0 месяцев), 54% были женского пола, 54% – представители европеоидной расы и 34% – представители азиатской расы.
Медиана возраста на момент набора в исследование составила 5.3 месяцев (2.2-6.9 месяцев), медиана времени между возникновением симптомов и приемом первой дозы составила 3.4 месяцев (1.0-6.0 месяцев).
На исходном уровне средний индекс по результатам теста детской больницы Филадельфии для оценки двигательных функций при нейромышечных заболеваниях у новорожденных (Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders, CHOP-INTEND) составил 22 балла (8.0-37.0) и средний индекс по результатам неврологической оценки младенцев по шкале Хаммерсмита, модуль 2 (Module 2 of the Hammersmith Infant Neurological Examination, HINE-2) составил 1.0 (0.0-5.0).
Первичной конечной точкой была доля пациентов с возможностью сидеть без поддержки в течение, как минимум, 5 секунд после 12 месяцев терапии (шкала крупной моторики BSID-III, пункт 22). Конечные точки по эффективности у пациентов, получавших препарат Эврисди®, сравнивали с таковыми в аналогичных группах нелеченных пациентов с манифестацией СМА в младенческом возрасте с естественным течением заболевания (критерии функционирования) – см. таблицу 1 ниже.
Таблица 1. Резюме ключевых результатов по эффективности на 12 месяце (FIREFISH часть 2).
| Конечные точки по эффективности | Доля пациентов N=41 (90% ДИ) |
| Основные критерии двигательной функции и развития | |
| BSID-III: способность сидеть без поддержки в течение, как минимум, 5 секунд Значение р на основании критерия функционирования 5%a | 29.3% (17.8%, 43.1%) <0.0001 |
| CHOP-INTEND: индекс 40 или выше Значение р на основании критерия функционирования 17%a | 56.1% (42.1%, 69.4%) <0.0001 |
| CHOP-INTEND: увеличение на ≥4 балла по сравнению с исходным уровнем Значение р на основании критерия функционирования 17%a | 90.2% (79.1%, 96.6%) <0.0001 |
| HINE-2: ответившие по основным критериям двигательной функцииb Значение р на основании критерия функционирования 12%a | 78.0% (64.8%, 88.0%) <0.0001 |
| Выживаемость и выживаемость без событий | |
| Выживаемость без событийc Значение р на основании критерия функционирования 42%a | 85.4% (73.4%, 92.2%) <0.0001 |
| Живы Значение р на основании критерия функционирования 60%a | 92.7% (82.2%, 97.1%) 0.0005 |
| Глотание и кормление | |
| Способность глотать | 85.4% (73.15%, 93.43%) |
| Способность принимать пищу через ротd | 82.9% (70.3%, 91.7%) |
| Использование ресурсов здравоохранения | |
| Отсутствие госпитализацийe | 48.8% (35.1%, 62.6%) |
a Значения р по выживаемости и выживаемости без событий основаны на Z-тестировании; значения р для других конечных точек (BSID-III, CHOP-INTEND, HINE-2) основаны на точном биноминальном тестировании. Доля выживаемости устанавливается с использованием методологии Каплана-Мейера.
b Согласно HINE-2: увеличение на ≥2 балла [или максимальный показатель] для способности брыкаться, ИЛИ увеличение на ≥1 балл по основным критериям двигательной функции: способность держать голову, перекатываться, сидеть, ползать, стоять или ходить И улучшение по большему числу категорий основных критериев двигательной функции по сравнению с ухудшением, что определяется как пациент, ответивший на этот анализ.
c Явлением считается достижение конечной точки по постоянной вентиляции легких (трахеостомия или ≥16 часов неинвазивной вентиляции легких в течение суток или интубация в течение >21 последовательного дня в отсутствие или после разрешения острого обратимого явления). Три пациента достигли конечной точки по постоянной вентиляции легких до 12 месяца. Все 3 пациента достигли увеличения, как минимум, на 4 балла по их индексу CHOP-1NTEND по сравнению с исходным уровнем.
d Включает пациентов, которых кормили исключительно через рот (всего 28 пациентов) и пациентов, которых кормили как через рот, так и с помощью трубки для кормления (всего 6 пациентов) на 12 месяце.
e Госпитализации включают все госпитализации, которые длились, как минимум, 2 дня.
После 12 месяцев терапии препаратом Эврисди® 29% (12/41) пациентов соответствовали критерию способности сидеть без поддержки (BS1D-III, пункт 22), 93% (38/41) пациентов были живы и 85% (35/41) пациентов были живы и без событий (без постоянной вентиляции), см. рисунок 1. Эти результаты указывают на клинически значимое отличие от естественного течения заболевания у нелеченных пациентов с манифестацией СМА в младенческом возрасте. Нелеченные пациенты с манифестацией СМА в младенческом возрасте никогда не смогут сидеть без поддержки, и ожидается, что только 25% пациентов смогут выжить без постоянной вентиляции легких после достижения возраста в 14 месяцев.
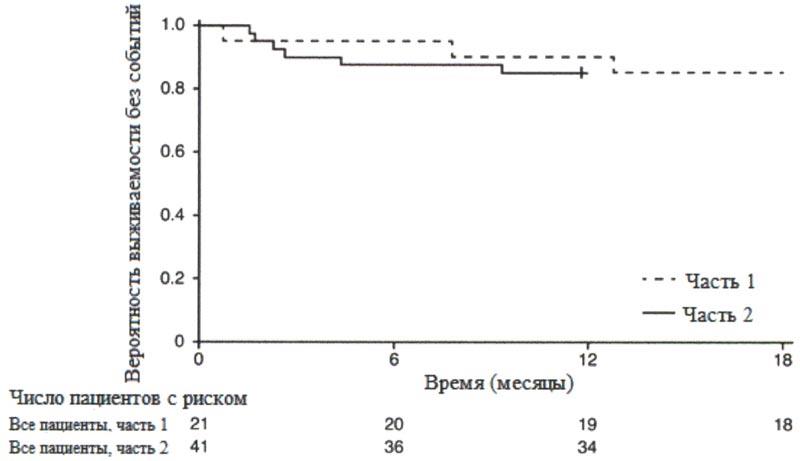 Рисунок 1. График Каплана-Мейера выживаемости без событий (FIREFISH часть 1 и часть 2).
Рисунок 1. График Каплана-Мейера выживаемости без событий (FIREFISH часть 1 и часть 2).+ цензурировано: один пациент в части 2 был цензурирован, поскольку он рано осуществил визит 12 месяца.
Большинство пациентов достигли ответа по категориям основных критериев двигательной функции HINE-2, включая определенный уровень способности держать голову (76%, 31/41), сидеть (61%, 25/41), перекатываться (56%, 23/41) и стоять (22%, 9/41). Согласно измерениям общего индекса CHOP-INTEND также наблюдались улучшения двигательной функции, см. рисунок 2.
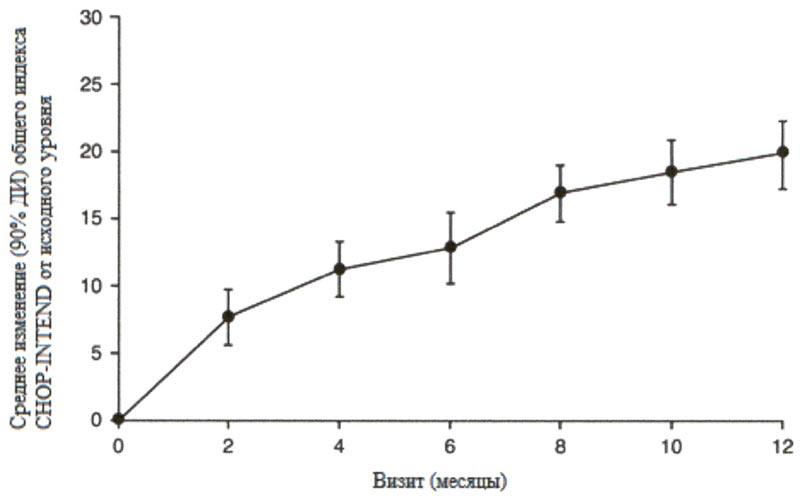 Рисунок 2. Среднее изменение по сравнению с исходным уровнем общего индекса СНОР-INTEND (FIREFISH часть 2).
Рисунок 2. Среднее изменение по сравнению с исходным уровнем общего индекса СНОР-INTEND (FIREFISH часть 2). FIREFISH часть 1
Эффективность препарата Эврисди® у пациентов со СМА 1 типа также подтверждается результатами из части 1 исследования FIREFISH. Для 21 пациента из части 1 характеристики на исходном уровне согласовывались с таковыми у пациентов с симптомами со СМА 1 типа.
Медиана возраста включения в исследование составила 6.7 месяцев (3.3-6.9 месяцев); медиана времени между возникновением симптомов и приемом первой дозы составила 4.0 месяцев (2.0-5.8 месяцев). Всего 17 пациентов получили терапевтическую дозу (дозу, выбранную для части 2) в течение первых 12 месяцев терапии. После 12 месяцев терапии 41% (7/17) пациентов могли сидеть без поддержки, как минимум, в течение 5 секунд (BSID-III, пункт 22). После 18 месяцев терапии 88% (15/17) пациентов были живы и без событий (без постоянной вентиляции), см. рисунок 1. Данные результаты по выживаемости и развитию двигательной функции согласовывались с результатами исследования FIREFISH часть 2.
Поздняя манифестация СМА
ВР39055 (SUNFISH) представляет собой многоцентровое исследование по изучению эффективности, безопасности, фармакокинетики и фармакодинамики препарата Эврисди® у пациентов со СМА 2 или 3 типа в возрасте 2-25 лет. Исследование состоит из двух частей. В части 1 осуществлялся подбор дозы. Часть 2 представляла собой рандомизированную, двойную слепую, плацебо-контролируемую, подтверждающую часть исследования. Пациенты из части 1 не принимали участия в части 2.
Первичной конечной точкой было изменение оценочного показателя двигательной функции – 32 (Motor Function Measure-32, MFM32) от исходного уровня на 12 месяце. Индекс MFM32 позволяет оценить обширный спектр двигательных функций у большого диапазона пациентов со СМА. Общий индекс MFM32 выражают в виде процента (0-100) от максимально возможного, причем более высокий показатель обозначают лучшую двигательную функцию. Индекс MFM32 измеряет двигательную функцию, в частности важные ежедневные двигательные способности. Небольшие изменения в двигательной функции могут привести к значительному приобретению или потере ежедневных двигательных способностей.
SUNFISH часть 2
SUNFISH часть 2 представляет собой рандомизированную, двойную слепую, плацебо-контролируемую часть исследования SUNFISH у 180 не амбулаторных пациентов со СМА 2 типа (71%) или 3 типа (29%). Пациентов рандомизировали в соотношении 2:1 в группу лечения препаратом Эврисди® в терапевтической дозе (см. раздел «Способ применения и дозы») или в группу плацебо. Рандомизация была стратифицирована по возрастным группам (2-5 лет, 6-11 лет, 12-17 лет и 18-25 лет).
Медиана возраста пациентов на момент начала лечения составляла 9.0 лет (2-25 лет), а медиана времени между возникновением первых симптомов СМА и приемом первой дозы составила 102.6 месяцев (1-275). Из 180 пациентов, включенных в исследование, 51% были женщинами, 67% – представителями европеоидной расы, и 19% – представителями азиатской расы.
На исходном уровне у 67% пациентов выявлен сколиоз (у 32% пациентов – тяжелый сколиоз). Среднее значение индекса MFM32 на исходном уровне составляло 46.1, а индекса по Обновленному модулю оценки функции верхних конечностей (Revised Upper Limb Module, RULM) – 20.1.
В целом, демографические характеристики пациентов на исходном уровне были хорошо сбалансированы между группами Эврисди® и плацебо, за исключением сколиоза (63.3% пациентов в группе препарата Эврисди® в сравнении с 73.3% пациентов в группе плацебо).
Первичный анализ изменения общего индекса MFM32 на 12 месяце по сравнению с исходным уровнем показал клинически и статистически значимое различие между пациентами, получавшими терапию препаратом Эврисди®, и пациентами, получавшими плацебо, в исследовании SUNFISH часть 2. Результаты первичного анализа и ключевых вторичных конечных точек представлены в таблице 2 и на рисунках 3 и 4.
Таблица 2. Резюме данных по эффективности у пациентов с поздней манифестацией СМА на 12 месяце лечения (SUNFISF1 часть 2).
| Конечная точка | Эврисди® (N=120) | Плацебо (N=60) |
| Первичная конечная точка | ||
| Изменение общего индекса MFM321 на 12 месяце от исходного уровня Среднее значение, рассчитанное по методу НК (95% ДИ) | 1.36 (0.61,2.11) | -0.19 (-1.22, 0.84) |
| Различие по сравнению с плацебо Расчетное значение (95% ДИ) Значение р2 | 1.55 (0.30, 2.81) 0.0156 | |
| Вторичные конечные точки | ||
| Доля пациентов с изменением общего индекса MFM321 на 12 месяце на ≥3 балла от исходного уровня (95% ДИ) | 38.3% (28.9, 47.6) | 23.7% (12.0, 35.4) |
| Отношение шансов для общего ответа (95% ДИ) Значение р3,4 с коррекцией (без коррекции) | 2.35 (1.01,5.44) 0.0469 (0.0469) | |
| Изменение общего индекса RULM5 на 12 месяце от исходного уровня Среднее значение, рассчитанное по методу НК (95% ДИ) | 1.61 (1.00, 2.22) | 0.02 (-0.83, 0.87) |
| Различие по сравнению с плацебо Расчетное (95% ДИ) значение р2,4 с коррекцией (без коррекции) | 1.59(0.55, 2.62) 0.0469 (0.0028) | |
НК = наименьшие квадраты
1 На основании правила по отсутствующим данным для индекса MFM32 6 пациентов были исключены из анализа (Эврисди® n = 115, контрольная группа плацебо n = 59).
2 Данные анализировали с использованием смешанной модели повторного измерения с общим индексом на исходном уровне, лечением, визитом, возрастной группой, лечением к визиту, а также исходным уровнем к визиту.
3 Данные анализировали с использованием логистической регрессии с общим индексом на исходном уровне, лечением и возрастной группой.
4 Значение р с коррекцией было получено для конечных точек, включенных в иерархическое тестирование, и выделялось на основании всех значений р, полученных для конечных точек в порядке увеличения иерархии до текущей конечной точки. Значение р без коррекции тестировали при уровне значимости 5%.
5 На основании правила отсутствующих данных для индекса RULM из анализа исключили 3 пациентов (Эврисди® n = 119; контрольная группа плацебо, n = 58).
При сравнении с группой плацебо пациенты, получавшие лечение препаратом Эврисди®, продемонстрировали значительное улучшение двигательной функции согласно оценке индекса MFM32 (среднее различие составило 1.55 баллов; р=0.0156) после 12 месяцев лечения. Пациенты в возрасте 2-5 лет, получавшие терапию препаратом Эврисди®, продемонстрировали наибольшее улучшение индекса MFM32 по сравнению с контрольной группой плацебо (увеличение на ≥3 балла у 78.1% в сравнении с 52.9%). Пациенты ≥18 лет, получавшие терапию препаратом Эврисди®, достигли стабилизации заболевания (изменение от исходного уровня по общему индексу MFM32 составило ≥0 баллов: 57.1% в сравнении с 37.5%).
У пациентов со СМА 2 типа и 3 типа, получавших терапию препаратом Эврисди®, наблюдалось сопоставимое улучшение по сравнению с исходным уровнем согласно индексу MFM32 (1.54 баллов [95% ДИ: 0.06, 3.02]; 1.49 баллов [95% ДИ: -0.94, 3.93] соответственно) по сравнению с контрольной группой плацебо.
Исследование также достигло вторичного независимого исхода двигательной функции, RULM. Согласно RULM отмечались статистически и клинически значимые улучшения в двигательной функции после 12 месяцев лечения по сравнению с исходным уровнем. Пациенты в возрасте 2-5 лет, получавшие лечение препаратом Эврисди®, продемонстрировали наибольшее улучшение индекса RULM (3.41 баллов [95% ДИ: 1.55, 5.26]), улучшение также отмечалось у пациентов ≥18 лет (1.74 баллов [95% ДИ: -1.06, 4.53]).
 Рисунок 3. Изменение среднего значения общего индекса MFM32 в течение 12 месяцев от исходного уровня (исследование SUNFISH часть 2)1.
Рисунок 3. Изменение среднего значения общего индекса MFM32 в течение 12 месяцев от исходного уровня (исследование SUNFISH часть 2)1. 1 Отклонение среднего значения, рассчитанного по методу НК, для изменения по сравнению с исходным уровнем в баллах по индексу MFM32 [95% ДИ].
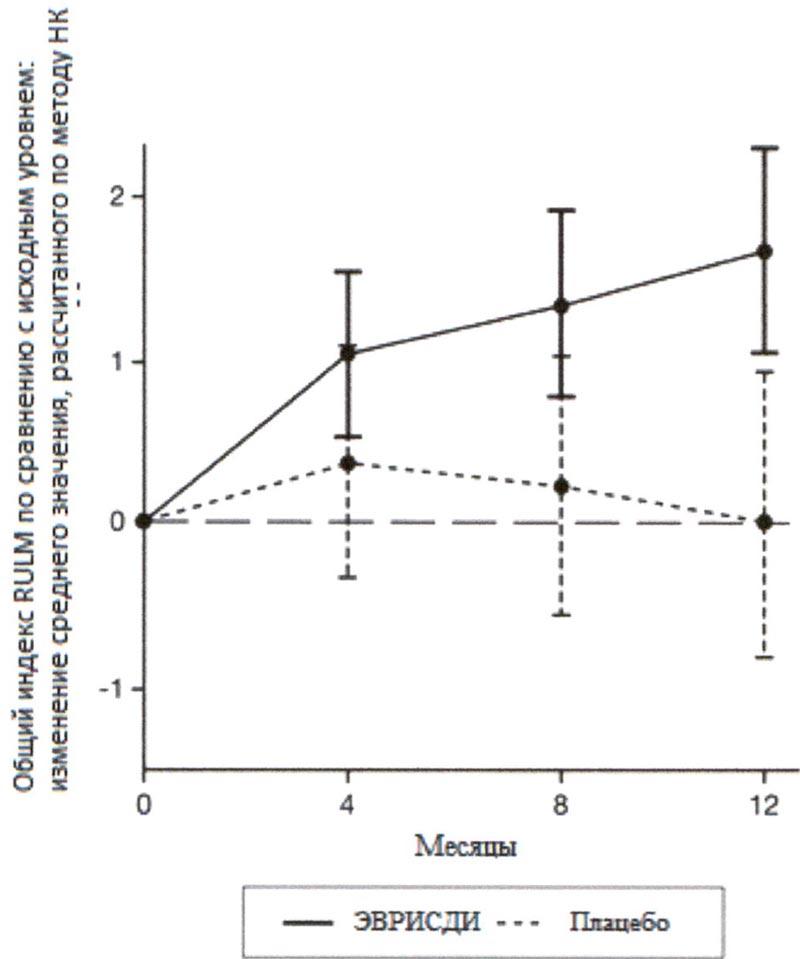 Рисунок 4. Изменение среднего значения общего индекса RULM в течение 12 месяцев от исходного уровня (SUNFISH часть 2)1.
Рисунок 4. Изменение среднего значения общего индекса RULM в течение 12 месяцев от исходного уровня (SUNFISH часть 2)1. 1 Отклонение среднего значения, рассчитанного по методу НК, для изменения по сравнению с исходным уровнем в баллах по индексу RULM [95% ДИ].
SUNFISH часть 1
Эффективность препарата Эврисди® у пациентов с поздней манифестацией СМА также подтверждается результатами из части 1 (часть исследования SUNFISH по подбору дозы). В часть 1 был включен 51 пациент со СМА 2 типа и 3 типа (включая 7 амбулаторных пациентов) в возрасте 2-25 лет.
После 1 года лечения препаратом в терапевтической дозе (доза, выбранная для части 2) было отмечено клинически значимое улучшение двигательной функции согласно измерениям индекса MFM32. Среднее изменение по сравнению с исходным уровнем составило 2.7 баллов (95% ДИ: 1.5, 3.8). Улучшение индекса MFM32 сохранялось ≤2 лет во время лечения препаратом Эврисди® (среднее изменение на 2.7 баллов [95% ДИ: 1.2, 4.2]).
В поисковом анализе двигательную функцию, оцениваемую согласно индексу MFM32, в части 1 исследования SUNFISH сравнивали с группой естественного течения СМА (на основании ключевых прогностических факторов). Общее изменение индекса MFM по сравнению с исходным уровнем после 1 года и 2 лет было выше у пациентов, получавших препарат Эврисди®, по сравнению с группой естественного течения заболевания (после 1 года: различие на 2.7 баллов; р <0.0001; после 2 лет: различие на 4.0 балла; р <0.0001). В группе естественного течения заболевания отмечалось ухудшение двигательной функции, как и ожидалось для естественного прогрессирования СМА (после 1 года: среднее изменение -0.6; после 2 лет: среднее изменение -2.0).
Применение у пациентов, ранее получавших лечение по поводу СМА
ВР39054 (JEWELFISH) представляет собой одногрупповое, открытое исследование по изучению безопасности, переносимости, фармакокинетики и фармакодинамики препарата Эврисди® у пациентов с манифестацией СМА в младенческом возрасте и у пациентов с поздней манифестацией СМА. В исследование были включены пациенты в возрасте 6 месяцев – 60 лет, которые ранее получали лечение по поводу СМА (включая нусинерсен и онасемноген абепарвовек). Из 174 пациентов, включенных в исследование, 76 пациентов ранее получали терапию нусинерсеном (9 пациентов со СМА 1 типа, 43 пациента со СМА 2 типа и 24 пациента со СМА 3 типа) и 14 пациентов ранее получали онасемноген абепарвовек (4 пациента со СМА 1 типа и 10 пациентов со СМА 2 типа). После 4 недель терапии препаратом Эврисди® у пациентов отмечалось увеличение уровня белка SMN в крови в среднем ≥2 раза выше по сравнению с исходным уровнем.
Фармакокинетика
Фармакокинетические параметры препарата Эврисди® были изучены у здоровых взрослых добровольцев и у пациентов со СМА.
После приема препарата Эврисди® в виде раствора внутрь фармакокинетика рисдиплама была приблизительно линейной между 0.6 и 18 мг. Фармакокинетика рисдиплама наилучшим образом описывалась с помощью популяционной фармакокинетической модели со всасыванием с тремя транзитными камерами, двухкамерным распределением и выведением первого порядка. Было обнаружено, что масса тела и возраст оказывают значимое влияние на фармакокинетику.
При применении рисдиплама в терапевтической дозе 0.2 мг/кг один раз в сутки у пациентов с манифестацией СМА в младенческом возрасте (от 2 до 7 месяцев при наборе в исследование) расчетная экспозиция (средняя площадь под кривой «концентрация-время» (AUC)0-24) составила 1930 нг.ч/мл. В исследовании SUNFISH (часть 2) при применении рисдиплама в терапевтической дозе 0.25 мг/кг один раз в сутки у пациентов с массой тела <20 кг и 5 мг один раз в сутки у пациентов с массой тела ≥20 кг (пациенты с поздней манифестацией СМА от 2 до 25 лет при наборе в исследование), расчетная экспозиция составила 2070 нг.ч/мл. Наблюдаемая максимальная концентрация (средняя Сmax) составила 194 нг.ч/мл при применении в дозе 0.2 мг/кг в исследовании F1REFISH и 120 нг.ч/мл в исследовании SUNFISH часть 2.
Всасывание
Рисдиплам быстро всасывался при приеме натощак, при этом время достижения максимальной концентрации (tmax) в плазме варьировало от 1 до 4 часов после приема внутрь. Прием пищи (высококалорийный завтрак с высоким содержанием жиров) не оказывал значимого влияния на экспозицию рисдиплама.
Распределение
Установленные популяционные фармакокинетические параметры составили 98 л для кажущегося центрального объема распределения, 93 л для периферического объема и 0.68 л/ч для межкамерного клиренса.
Рисдиплам преимущественно связывался с сывороточным альбумином без какого-либо связывания с альфа-1-кислым гликопротеином, свободная фракция составила 11%.
Метаболизм
Рисдиплам в основном метаболизируется флавин-монооксигеназой 1 и 3 (FMO 1 и 3), а также изоферментами цитохрома (CYP) 1А1, 2J2, 3А4 и 3А7.
Одновременный прием итраконазола, мощного ингибитора CYP3A, в дозе 200 мг два раза в сутки с однократным пероральным приемом рисдиплама в дозе 6 мг не выявил клинически значимого влияния на фармакокинетику рисдиплама (показатель AUC повышен на 11%, показатель Сmax снижен на 9%).
Выведение
В ходе популяционного фармакокинетического анализа был установлен кажущийся клиренс (CL/F) рисдиплама 2.6 л/ч.
Эффективный период полувыведения рисдиплама составил приблизительно 50 часов у пациентов со СМА.
Рисдиплам не является субстратом белка множественной лекарственной устойчивости 1 (MDR1) у человека.
Приблизительно 53% дозы (14% неизменного рисдиплама) выводилось с калом и 28% с мочой (8% неизменного рисдиплама). Исходный препарат был основным компонентом в плазме, что составило 83% от общих компонентов препарата, находящихся в кровотоке. Фармакологически неактивный метаболит М1 был определен как основной циркулирующий метаболит.
Особые группы пациентов
Пациенты детского возраста
Масса тела и возраст были определены как ковариаты в популяционном фармакокинетическом анализе. Таким образом, доза определяется в зависимости от возраста (младше и старше 2 лет) и массы тела (до 20 кг) для достижения сходных экспозиций среди пациентов всех возрастов и с различной массой тела. Данные у пациентов младше 2 месяцев отсутствуют.
Пациенты пожилого возраста
Специальных исследований по изучению фармакокинетики препарата Эврисди® у пациентов со СМА старше 60 лет не проводилось. Пациенты со СМА ≤60 лет принимали участие в исследовании JEWELFISH. Субъекты без СМА ≤69 лет принимали участие в клинических исследованиях по фармакокинетике. Согласно результатам данных исследований коррекции дозы у пациентов ≤69 лет не требуется.
Пациенты с нарушением функции почек
Исследований по изучению фармакокинетики рисдиплама у пациентов с нарушением функции почек не проводилось. Рисдиплам в виде неизменного соединения выводится почками в малой степени (8%).
Пациенты с нарушением функции печени
Нарушение функции печени легкой или средней степени тяжести не оказывало влияния на фармакокинетику рисдиплама. После применения 5 мг рисдиплама средние соотношения для Сmax и AUC составили 0.95 и 0.80 у субъектов с нарушением функции печени легкой степени тяжести (n=8), а также 1.20 и 1.08 у субъектов с нарушением функции печени средней степени тяжести (п=8) по сравнению с соответствующими здоровыми добровольцами (n=10). Безопасность и фармакокинетика у пациентов с нарушением функции печени тяжелой степени тяжести не изучались.
Этническая принадлежность
Фармакокинетика рисдиплама у японских субъектов и субъектов европеоидной расы не отличалась.
Показания к применению
Лечение спинальной мышечной атрофии (СМА) у взрослых и детей с 2 месяцев.
Противопоказания
Повышенная чувствительность к рисдипламу или к другим вспомогательным веществам препарата в анамнезе.
Беременность и период грудного вскармливания.
Младенцы младше 2 месяцев.
Применение при беременности и в период грудного вскармливания
Беременность
Применение препарата Эврисди® при беременности противопоказано.
Данные клинических исследований препарата Эврисди® у беременных женщин отсутствуют. Была показана эмбриофетальная токсичность и тератогенность рисдиплама у животных. На основании данных, полученных в исследованиях у животных, установлено, что рисдиплам проходит через плацентарный барьер и может оказывать повреждающее действие на плод.
Безопасность применения препарата Эврисди® во время родов и родоразрешения не установлена.
Период грудного вскармливания
Применение препарата в период грудного вскармливания противопоказано. Неизвестно, выводится ли препарат Эврисди® с грудным молоком у человека. Исследования у крыс показали, что рисдиплам выводится с грудным молоком.
Фертильность
Пациенты-мужчины
Согласно данным доклинических исследований фертильность у мужчин может быть нарушена в ходе терапии препаратом Эврисди®. В репродуктивных органах крыс и обезьян наблюдались дегенеративные формы и снижение числа сперматозоидов. Влияние на сперматозоиды обратимо после отмены рисдиплама.
Перед началом лечения препаратом Эврисди® с пациентами-мужчинами необходимо обсудить стратегии сохранения фертильности. Пациенты-мужчины могут рассмотреть возможность консервации спермы перед началом лечения или по прошествии минимум 4 месяцев после завершения лечения. Пациенты-мужчины, которые хотят завести ребенка, должны прекратить лечение препаратом Эврисди®, как минимум, на 4 месяца. Лечение может быть продолжено после зачатия.
Пациенты-женщины
На основании данных доклинических исследований влияния препарата Эврисди® на фертильность у женщин не ожидается.
Тестирование на беременность
Перед началом терапии препаратом Эврисди® следует проверить статус беременности у женщин репродуктивного потенциала. Беременных женщин следует четко проинформировать о потенциальном риске для плода.
Контрацепция
Пациенты (мужчины и женщины) репродуктивного потенциала должны соблюдать следующие требования в отношении контрацепции:
- пациенты-женщины детородного потенциала должны использовать высокоэффективные методы контрацепции во время лечения и в течение, как минимум, 1 месяца после приема последней дозы;
- пациенты-мужчины и их партнерши детородного потенциала должны вместе использовать высокоэффективные методы контрацепции во время лечения и в течение, как минимум, 4 месяцев после приема последней дозы пациентом-мужчиной.
Способ применения и дозы
Раствор для приема внутрь должен быть приготовлен медицинским работником перед его выдачей пациенту.
Общие рекомендации
Лечение СМА следует начать как можно раньше после постановки диагноза.
Препарат Эврисди® принимают внутрь один раз в сутки приблизительно в одно и то же время каждый день с помощью предоставляемого перорального шприца.
Рекомендуемая суточная доза препарата Эврисди® у пациентов со СМА определяется в зависимости от возраста и массы тела (см. таблицу 3).
Таблица 3. Режим дозирования в зависимости от возраста и массы тела.
| Возраст и масса тела | Рекомендуемая суточная доза |
| от 2 месяцев до <2 лет | 0.20 мг/кг |
| ≥2 лет (масса тела <20 кг) | 0.25 мг/кг |
| ≥2 лет (масса тела ≥20 кг) | 5 мг |
Изменения дозы должны проводиться под контролем медицинского работника. Терапия препаратом в дозах выше 5 мг в сутки не изучалась. Данные у младенцев младше 2 месяцев отсутствуют.
Способ применения
Для приема суточной дозы препарата Эврисди® следует использовать предоставляемый многоразовый пероральный шприц. Медицинский работник перед началом лечения должен обсудить с пациентом или лицом, осуществляющим уход за пациентом, как подготовить назначенную суточную дозу для приема.
Пациенту необходимо выпить воду после приема препарата Эврисди® для гарантии, что препарат проглочен полностью. В случае, если пациент не может глотать и у него установлена назогастральная или гастростомическая трубка, следует ввести препарат через трубку. После введения препарата Эврисди® необходимо промыть трубку водой.
Задержка введения или пропуск дозы
Препарат Эврисди® принимают внутрь один раз в сутки приблизительно в одно и то же время каждый день.
Если прием препарата Эврисди® был пропущен и прошло не более 6 часов с момента запланированного приема, следует принять препарат как можно скорее.
Если прошло более 6 часов с момента запланированного приема, следует пропустить прием и принять препарат в обычное запланированное время на следующий день.
Если препарат не был проглочен полностью или после его приема произошла рвота, не следует принимать препарат повторно для восполнения неполной дозы. Необходимо дождаться следующего дня и принять препарат в обычное запланированное время.
Дозирование в особых случаях
Пациенты детского возраста
Эффективность и безопасность препарата Эврисди® у пациентов <2 месяцев еще не установлена (см. раздел «Фармакологические свойства», подраздел «Клиническая эффективность»).
Пациенты пожилого возраста
Фармакокинетика и безопасность препарата Эврисди® оценивались у субъектов без СМА в возрасте ≤69 лет. Применение препарата Эврисди® у пациентов со СМА старше 60 лет не изучалось (см. раздел «Фармакологические свойства», подраздел «Особые группы пациентов»).
Пациенты с нарушением функции почек
Эффективность и безопасность препарата Эврисди® у пациентов с нарушением функции почек не изучались. Не ожидается необходимости в коррекции дозы у пациентов с нарушением функции почек (см. раздел «Фармакологические свойства», подраздел «Особые группы пациентов»).
Пациенты с нарушением функции печени
Коррекции дозы у пациентов с нарушением функции печени легкой или средней степени тяжести не требуется. Применение препарата Эврисди® у пациентов с нарушением функции печени тяжелой степени тяжести не изучалось (см. раздел «Фармакологические свойства», подраздел «Особые группы пациентов» и раздел «Особые указания»).
Особая информация об использовании, обращении и утилизации препарата
Приготовление раствора препарата Эврисди® из порошка осуществляется медицинским работником перед выдачей пациенту.
Приготовление раствора для приема внутрь 0.75 мг/мл из 60 мг порошка (см. в конце инструкции Руководство по разведению – только для медицинских работников).
Следует соблюдать осторожность при обращении с порошком препарата Эврисди® для приготовления раствора для приема внутрь (см. раздел «Особые указания»). Следует избегать вдыхания и прямого контакта сухого порошка и приготовленного раствора с кожей или слизистыми оболочками. Следует использовать одноразовые перчатки во время разведения и при очистке наружной поверхности флакона/крышки и рабочей поверхности после разведения.
Если контакт все же произошел, нужно тщательно промыть данный участок водой с мылом, а глаза – просто водой.
Выбор перорального шприца для применения назначенной суточной дозы препарата Эврисди®
Таблица 4. Выбор перорального шприца для применения назначенной суточной дозы.
| Размер шприца | Объем дозы | Деления на шприце |
| 6 мл | 1.0 мл-6.0 мл | 0.1 мл |
| 12 мл | 6.2 мл-6.6 мл | 0.2 мл |
При расчете объема дозы следует принять во внимание деления на шприце. Необходимо округлить объем дозы до ближайшего деления, отмеченного на выбранном пероральном шприце.
Пациенты должны принять препарат Эврисди® незамедлительно после его набора в пероральный шприц. Если препарат не был принят в течение 5 минут, следует утилизировать раствор препарата из перорального шприца и набрать новую дозу.
Несовместимость
Не обнаружено признаков несовместимости между препаратом Эврисди® и рекомендуемыми пероральными шприцами.
Инструкции по уничтожению неиспользованного препарата или препарата с истекшим сроком годности
Попадание лекарственного препарата в окружающую среду должно быть сведено к минимуму. Не следует утилизировать препарат с помощью сточных вод или вместе с бытовыми отходами. Уничтожение неиспользованного препарата или препарата с истекшим сроком годности должно проводиться в соответствии с локальными требованиями.
Руководство по использованию препарата
Пациенты/лица, осуществляющие уход за пациентами, должны ознакомиться с важной информацией касательно приема препарата, которая представлена ниже.
Важная информация для пациентов/лиц, осуществляющих уход за пациентами, по препарату Эврисди®
- Попросите медицинского работника показать Вам правильный шприц, который Вы должны использовать, и как отмерять назначенную суточную дозу.
- Всегда используйте многоразовые пероральные шприцы, предоставляемые в упаковке, для того, чтобы отмерять назначенную Вам суточную дозу. Пероральные шприцы защищают препарат от света.
- Предоставляется по два пероральных шприца каждого размера на случай, если один из них будет утерян или поврежден. Если оба пероральных шприца утеряны или повреждены, свяжитесь с медицинским работником, который проконсультирует Вас, как можно продолжить применение препарата.
- См. выше подраздел «Выбор перорального шприца для применения назначенной суточной дозы», чтобы выбрать правильный пероральный шприц для использования. Проконсультируйтесь с медицинским работником в случае возникновения вопросов по выбору правильного перорального шприца.
- Не применяйте препарат Эврисди®, если во флаконе отсутствует адаптер для флакона, и свяжитесь с медицинским работником.
- Не применяйте препарат Эврисди® последаты утилизации, указанной на этикетке флакона. Если на этикетке флакона отсутствует дата утилизации, свяжитесь с медицинским работником.
- Не смешивайте препарат Эврисди® с пищей и напитками.
- Не используйте препарат Эврисди®, если флакон или пероральные шприцы повреждены.
- Избегайте контакта препарата Эврисди® с кожей. Если это произошло, промойте данный участок водой с мылом.
- Если Вы разлили препарат Эврисди®, протрите данную область сухим бумажным полотенцем и промойте водой с мылом. Бумажное полотенце следует выбросить в мусор и хорошо вымыть руки водой с мылом.
- Если во флаконе осталось количество препарата, недостаточное для приема назначенной дозы, утилизируйте флакон с оставшимся препаратом и использованные пероральные шприцы в соответствии с локальными требованиями; используйте новый флакон с препаратом Эврисди® для приема назначенной дозы. Не смешивайте препарат Эврисди® из нового флакона с препаратом Эврисди® из используемого на данный момент флакона.
Хранение раствора препарата Эврисди®
Следует хранить приготовленный раствор препарата в оригинальном флаконе темного стекла, плотно закрытом крышкой, в холодильнике (при температуре 2-8 °С) в вертикальном положении, в течение до 64 дней после разведения. Не замораживать.
А) Подготовка и отбор назначенной дозы
Как выбрать правильный пероральный шприц для применения назначенной дозы препарата Эврисди®
Если назначенный объем суточной дозы составляет от 1 мл до 6 мл, следует использовать пероральный шприц на 6 мл (с серой этикеткой).
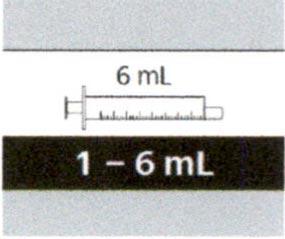
Если назначенный объем суточной дозы составляет от 6.2 мл и выше, следует использовать пероральный шприц на 12 мл (с коричневой этикеткой).

Как подготовить назначенный объем дозы препарата Эврисди®
Шаг А1
Снимите крышку, нажав на нее вниз и затем повернув налево (против часовой стрелки). Не выбрасывайте крышку.
Шаг А2
Нажимайте на поршень перорального шприца до конца для удаления воздуха.
Шаг А3
Держа флакон вертикально, вставьте кончик шприца в адаптер для флакона.
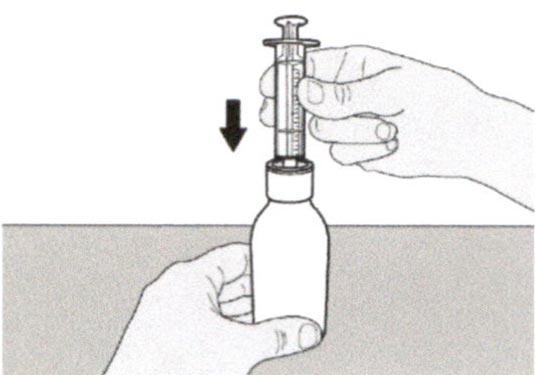
Шаг А4
Аккуратно переверните флакон вместе с пероральным шприцем, кончик которого плотно вставлен в адаптер для флакона.
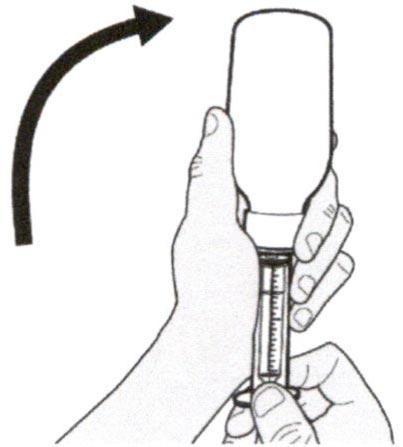
Шаг А5
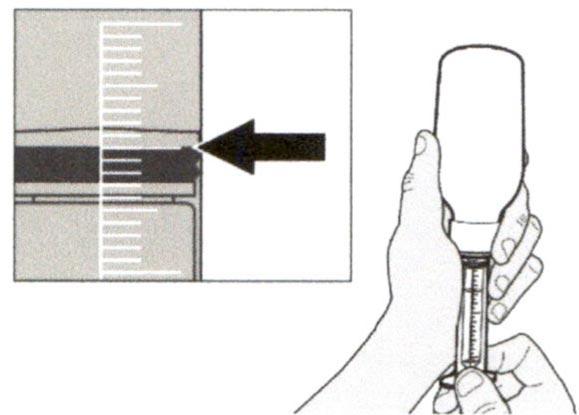
Медленно оттягивайте поршень, чтобы набрать назначенный объем дозы препарата Эврисди®.
Верхняя поверхность черного ограничителя поршня должна находиться на одной линии с делением (мл) на пероральном шприце, соответствующем назначенному объему суточной дозы.
После того, как доза была набрана правильно, удерживайте поршень на месте и не давайте ему смещаться.
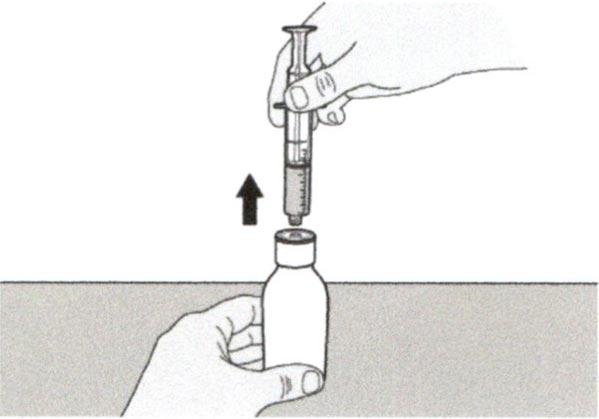
Шаг А6
Продолжайте удерживать поршень на месте и не давайте ему смещаться. Оставьте пероральный шприц в адаптере для флакона и поверните флакон вертикально, горлышком вверх. Поместите флакон на плоскую поверхность. Удалите пероральный шприц из адаптера для флакона, аккуратно вытягивая прямо вверх пероральный шприц.
Шаг А7
Удерживайте пероральный шприц, чтобы его кончик был направлен вверх. Проверьте препарат в пероральном шприце. Если в растворе присутствуют большие пузырьки воздуха или если Вы неправильно набрали дозу препарата Эврисди®, плотно вставьте кончик шприца в адаптер для флакона. Нажимайте на поршень шприца до конца, чтобы переместить препарат обратно во флакон и повторите шаги А4-А7.
Примите или введите препарат Эврисди® незамедлительно после его набора в пероральный шприц.
Если препарат не был принят в течение 5 минут, следует утилизировать раствор препарата из перорального шприца и набрать новую дозу.
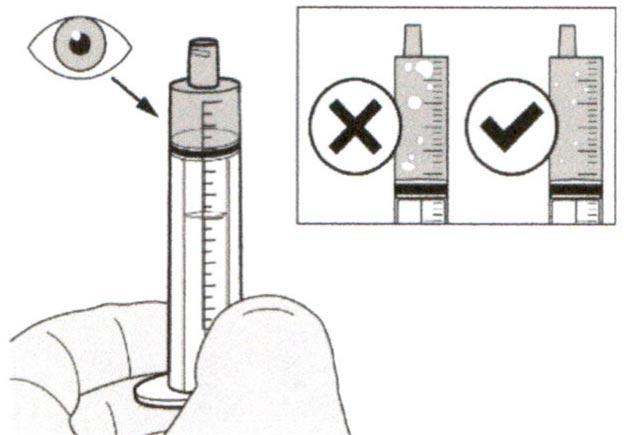
Шаг А8
Наденьте крышку обратно на флакон. Поверните крышку вправо (по часовой стрелке), чтобы плотно закрыть флакон.
Не удаляйте адаптер для флакона из флакона.
Если Вы принимаете препарат Эврисди® перорально, см. пункт Б)
Прием препарата Эврисди® перорально.
Если Вы вводите препарат Эврисди® через гастростомическую трубку, см. пункт В)
Введение препарата Эврисди® через гастростомическую трубку.
Если Вы вводите препарат Эврисди® через назогастральную трубку, см. пункт Г)
Введение препарата Эврисди® через назогастральную трубку.
Б) Прием препарата Эврисди® перорально
Следует сесть прямо для перорального приема препарата Эврисди®.
Шаг Б1
Поместите пероральный шприц в рот, расположив кончик шприца вдоль любой щеки.
Медленно нажимайте на поршень до конца для введения полной дозы препарата Эврисди®.
Введение препарата Эврисди® прямо в горло или слишком быстро может привести к удушью.
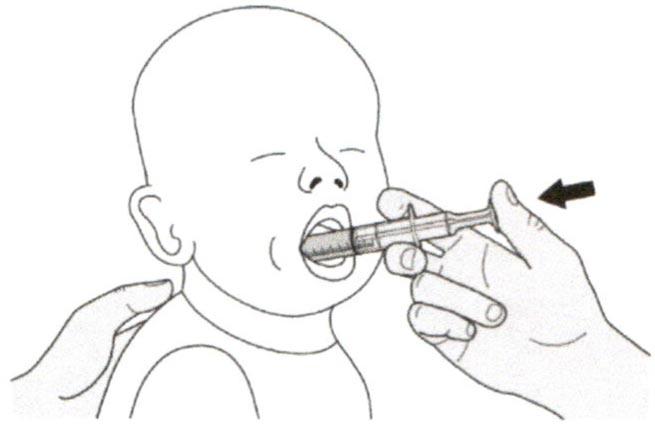
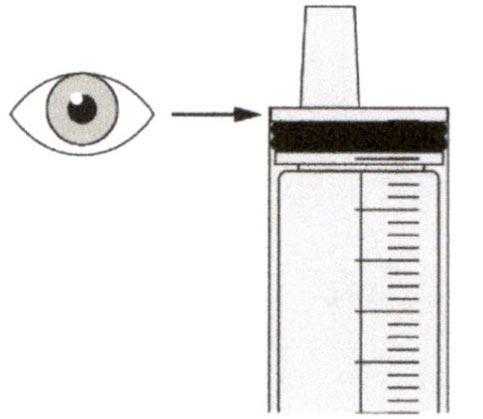
Шаг Б2
Проверьте, что в пероральном шприце не остался раствор препарата.
Шаг Б3
Выпейте воду сразу после приема назначенной дозы препарата Эврисди®.
Затем см. Шаг Д по очистке шприца.
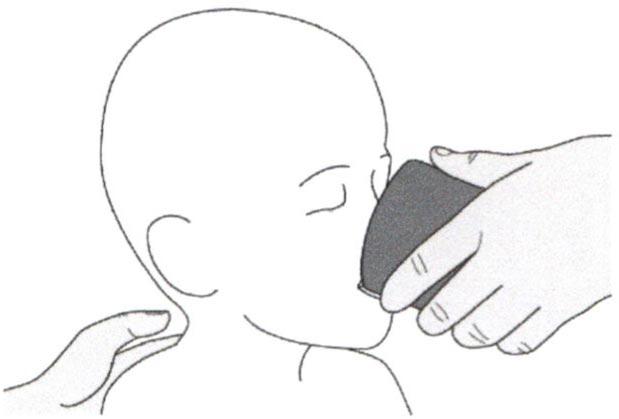
В) Введение препарата Эврисди® через гастростомическую трубку
Если Вы вводите препарат Эврисди® через гастростомическую трубку, проконсультируйтесь с медицинским работником касательно проверки гастростомической трубки перед введением препарата Эврисди®.
Шаг В1
Поместите кончик перорального шприца в гастростомическую трубку.
Медленно нажимайте на поршень до конца для введения полной дозы препарата Эврисди®.
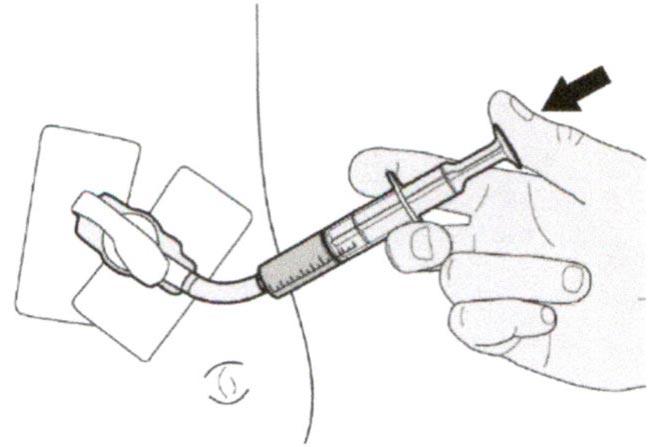
Шаг В2
Проверьте, что в пероральном шприце не остался раствор препарата.
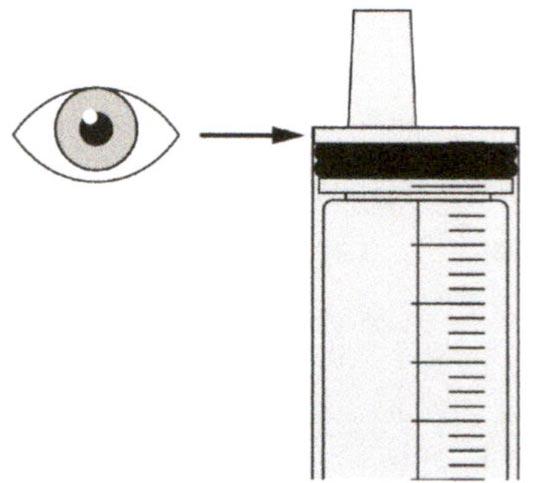
Шаг В3
Сразу после введения назначенной дозы препарата Эврисди® следует промыть гастростомическую трубку 10-20 мл воды.
Затем см. Шаг Д по очистке шприца.
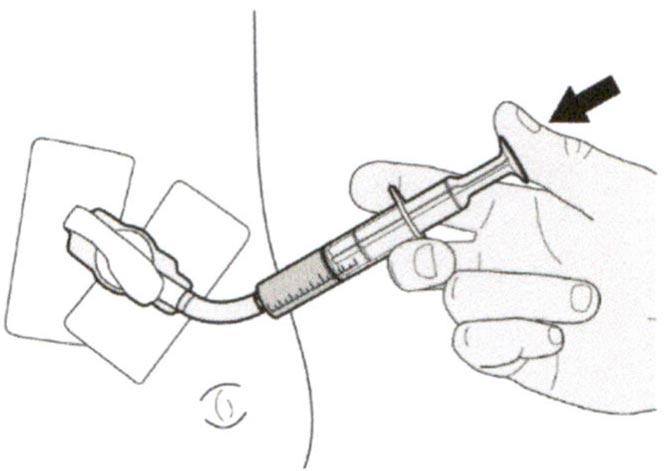
Г) Введение препарата Эврисди® через назогастральную трубку
Если Вы вводите препарат Эврисди® через назогастральную трубку, проконсультируйтесь с медицинским работником касательно проверки назогастральной трубки перед введением препарата Эврисди®.
Шаг Г1
Поместите кончик перорального шприца в назогастральную трубку.
Медленно нажимайте на поршень до конца для введения полной дозы препарата Эврисди®.
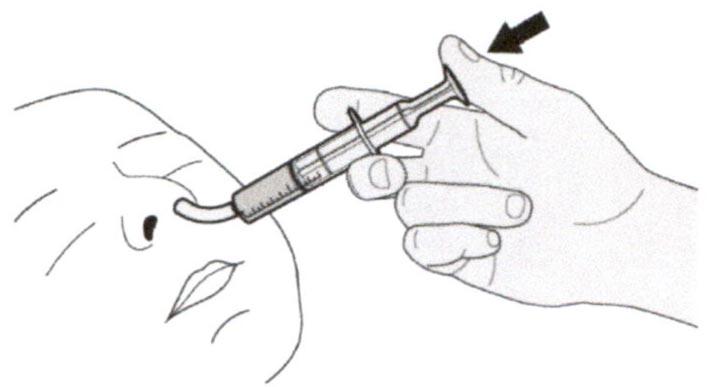
Шаг Г2
Проверьте, что в пероральном шприце не остался раствор препарата.
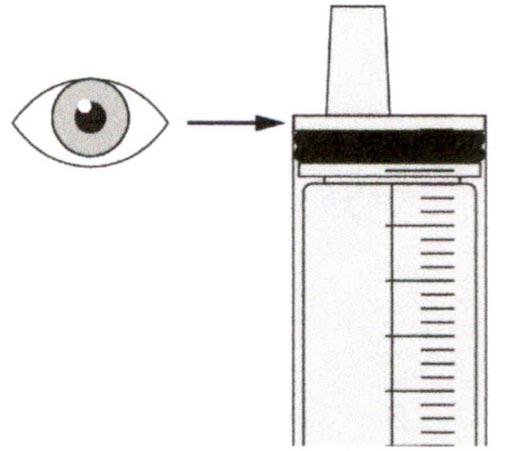
Шаг Г3
Сразу после введения назначенной дозы препарата Эврисди® следует промыть назогастральную трубку 10-20 мл воды.
Затем см. Шаг Д по очистке шприца.
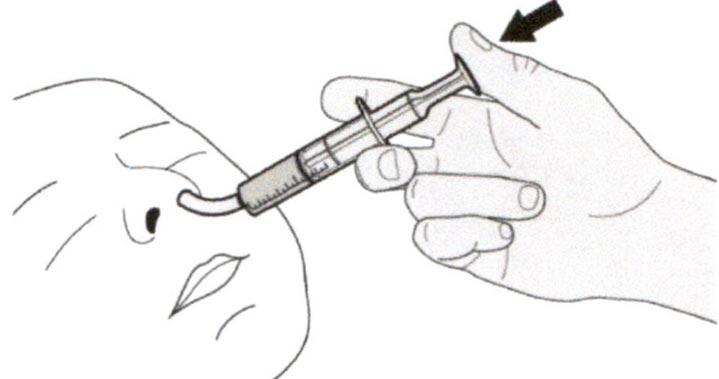
Д) Очистка перорального шприца после использования
Шаг Д1
Удалите поршень из перорального шприца.
Хорошо промойте цилиндр перорального шприца чистой водой.
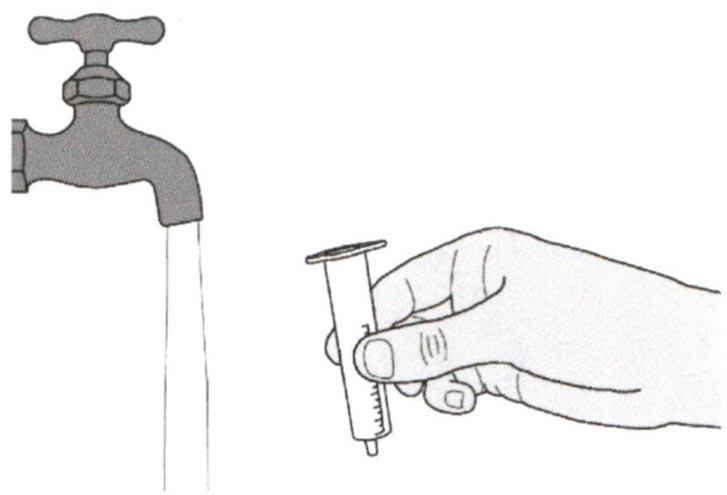
Шаг Д2
Хорошо промойте поршень перорального шприца чистой водой.
Шаг Д3
Убедитесь, что цилиндр и поршень перорального шприца чистые.
Поместите цилиндр и поршень перорального шприца на чистую поверхность в безопасном месте, чтобы они высохли.
Вымойте руки.
После того как цилиндр и поршень перорального шприца высохнут, вставьте поршень в цилиндр перорального шприца и храните вместе с препаратом для дальнейшего использования.
Побочное действие
Резюме профиля безопасности
Профиль безопасности препарата Эврисди® основан на трех клинических исследованиях FIREFISH, SUNFISH и JEWELFISH.
FIREFISH представляет собой открытое исследование, состоящее из двух частей, в которое включено 62 пациента с манифестацией СМА в младенческом возрасте (от 2.2 до 6.9 месяцев). Пятьдесят пять пациентов получали терапию препаратом Эврисди® в течение >12 месяцев (см. раздел «Фармакологические свойства», подраздел «Клиническая эффективность»). Нежелательные реакции, которые наблюдались в клинических исследованиях у пациентов с манифестацией СМА в младенческом возрасте, представленные в таблице 5 ниже, основаны на данных объединенного анализа у пациентов из частей 1 и 2 клинического исследования FIREFISH. Нежелательные реакции определяются как нежелательные явления, которые отмечаются у ≥5% пациентов и для которых возможна причинно-следственная связь с препаратом Эврисди®.
SUNFISH представляет собой исследование, состоящее из двух частей, в которое включены пациенты с поздней манифестацией СМА (от 2 до 25 лет) (см. раздел «Фармакологические свойства», подраздел «Клиническая эффективность»).
Нежелательные реакции, которые наблюдались в клинических исследованиях у пациентов с поздней манифестацией СМА, основаны на данных исследования SUNFISH часть 2 (n=180), рандомизированной, двойной слепой, плацебо-контролируемой части исследования с периодом последующего наблюдения, как минимум, 12 месяцев. Нежелательные реакции определяются как нежелательные явления, которые отмечаются на ≥5% чаще или, как минимум, в 2 раза чаще по сравнению с пациентами, получавшими плацебо, и для которых возможна причинно-следственная связь с препаратом Эврисди®.
Таблица 5. Резюме нежелательных реакций у пациентов с манифестацией СМ А в младенческом возрасте в исследовании FIREFISH (части 1 и 2).
| Класс систем органов | Нежелательная реакция | Частота N=62 n (%) | Число явлений/ 100 пациенто-лет Общая экспозиция пациенто-лет = 87.9 | Категория частоты |
| Нарушения со стороны желудочно-кишечного тракта | Диарея | 10 (16.1) | 13.7 | Очень часто |
| Нарушения со стороны кожи и подкожных тканей | Сыпь* | 17 (27.4) | 23.9 | Очень часто |
* Включая сыпь, макулопапулезную сыпь, эритему, дерматит, аллергический дерматит, папулезную сыпь, фолликулит.
Таблица 6. Резюме нежелательных реакций у пациентов с поздней манифестацией СМА в исследовании SUNFISH часть 2.
| Класс систем органов | Нежелательная реакция | Частота N=120 n (%) | Плацебо N=60 n (%) | Категория частоты |
| Нарушения со стороны желудочно-кишечного тракта | Диарея | 20 (16.7) | 5 (8.3%) | Очень часто |
| Нарушения со стороны кожи и подкожных тканей | Сыпь* | 20 (16.7) | 1 (1.7) | Очень часто |
* Включая сыпь, макулопапулезную сыпь, эритему, аллергический дерматит, эритематозную сыпь, фолликулит, папулезную сыпь.
Нежелательные реакции диарея и сыпь отмечались без определенного времени или клинической картины и разрешались несмотря на продолжающееся лечение препаратом Эврисди® у пациентов с манифестацией СМА в младенческом возрасте и у пациентов с поздней манифестацией СМА. Данные явления не указывают на влияние на эпителиальные ткани, которые отмечались в исследованиях у животных.
Профиль безопасности у пациентов, ранее получавших терапию по поводу СМА
Профиль безопасности препарата Эврисди® у пациентов, ранее получавших терапию по поводу СМА в исследовании JEWELFISH, сопоставим с таковым у пациентов, ранее не получавших лечения в исследованиях FIREFISH (часть 1 и 2) и SUNFISH (часть 1 и 2). В исследовании JEWELFISH принимали участие 76 пациентов, которые ранее получали нусинерсен и 14 пациентов, которые ранее получали онасемноген абепарвовек (см. раздел «Фармакологические свойства», подраздел «Клиническая эффективность»).
Передозировка
Случаев передозировки препарата Эврисди® в клинических исследованиях не было.
Лечение
Известного антидота для применения в случае передозировки препарата Эврисди® нет.
В случае передозировки следует тщательно наблюдать за пациентом и провести поддерживающую терапию.
Взаимодействие с другими лекарственными средствами
Рисдиплам в основном метаболизируется флавин-монооксигеназой 1 и 3 (FMO 1 и 3), а также изоферментами цитохрома (CYP) 1А1, 2J2, ЗА4 и ЗА7. Рисдиплам не является субстратом белка множественной лекарственной устойчивости 1 (MDR1) у человека.
Влияние других лекарственных средств на препарат Эврисди®
Одновременный прием итраконазола, мощного ингибитора CYP3A, в дозе 200 мг два раза в сутки с однократным пероральным приемом рисдиплама в дозе 6 мг не выявил клинически значимого влияния на фармакокинетику рисдиплама (показатель AUC повышен на 11%, показатель Сmax снижен на 9%). При одновременном применении ингибитора CYP3A и препарата Эврисди® коррекции дозы последнего не требуется.
Лекарственного взаимодействия с препаратами, метаболизирующимися посредством флавин-монооксигеназы 1 и 3 (FMO 1 и 3), не ожидается.
Влияние препарата Эврисди® на другие лекарственные средства
В условиях in vitro рисдиплам и его основной циркулирующий метаболит М1 не индуцировали CYP1A2, 2В6, 2С8, 2С9, 2С19 или 3А4. В условиях in vitro рисдиплам и M1 не ингибировали (обратимо или в зависимости от времени) какой-либо из изучаемых ферментов CYP (CYP1A2, 2В6, 2С8, 2С9, 2С19, 2D6) за исключением CYP3A.
Препарат Эврисди® является слабым ингибитором CYP3A. При введении препарата Эврисди® здоровым взрослым добровольцам один раз в сутки в течение двух недель экспозиция мидазолама (чувствительного субстрата CYP3A) незначительно повышалась (AUC 11%, Сmax 16%). Степень взаимодействия не считается клинически значимой, и, таким образом, коррекции дозы субстратов CYP3A не требуется. Согласно фармакокинетическому моделированию, основанному на физиологии, ожидается, что данный эффект будет иметь сходную выраженность у младенцев старше 2 месяцев и детей.
Исследования in vitro показали, что рисдиплам и его основной метаболит не являются значимыми ингибиторами MDR1, транспортного полипептида органических анионов ОАТР1В1 и ОАТР1В3 и переносчика органических анионов ОАТ1 и ОАТ3 у человека.
Рисдиплам и его метаболит, однако, являются ингибиторами переносчика органических катионов ОСТ2, белка экструзии лекарственных препаратов и токсинов МАТЕ1 и МАТЕ2-К в условиях in vitro. При терапевтических концентрациях препарата взаимодействий с субстратами ОСТ2 не ожидается. Согласно данным, полученным в условиях in vitro, при применении препарата Эврисди® плазменные концентрации препаратов, которые выводятся посредством МАТЕ1 и МАТЕ2-К, могут повышаться. Клиническая значимость одновременного применения с субстратами МАТЕ1/2-К неизвестна.
Особые указания
Эмбриофетальная токсичность
При изучении применения препарата у животных наблюдалась эмбриофетальная токсичность. Пациенты репродуктивного потенциала должны быть проинформированы о рисках и использовать высокоэффективные методы контрацепции во время лечения и в течение, как минимум, 1 месяца после приема последней дозы препарата Эврисди® пациентом-женщиной и в течение, как минимум, 4 месяцев после приема последней дозы препарата Эврисди® пациентом-мужчиной (см. раздел «Применение при беременности и в период грудного вскармливания»).
Возможное влияние на фертильность у мужчин
Обратимое влияние препарата Эврисди® на мужскую фертильность было показано в исследовании у животных. В связи с этим, пациенты-мужчины не должны быть донорами спермы во время лечения и в течение 4 месяцев после приема последней дозы препарата Эврисди® (см. раздел «Применение при беременности и в период грудного вскармливания»).
Нарушение функции печени
Фармакокинетику, безопасность и переносимость рисдиплама в дозе 5 мг оценивали у субъектов с нарушением функции печени легкой или средней степени тяжести в специальном клиническом исследовании.
Нарушение функции печени легкой или средней степени тяжести не оказывало влияния на фармакокинетику рисдиплама. Таким образом, пациентам с нарушением функции печени легкой или средней степени тяжести коррекции дозы не требуется. Применение препарата Эврисди® у пациентов с нарушением функции печени тяжелой степени тяжести не изучалось (см. раздел «Фармакологические свойства», подраздел «Особые группы пациентов» и раздел «Способ применения и дозы», подраздел «Дозирование в особых случаях»).
Злоупотребление приемом препарата или лекарственная зависимость
Препарат Эврисди® не обладает потенциалом, который может привести к злоупотреблению приемом препарата и лекарственной зависимости.
Влияние на способность управлять транспортными средствами, механизмами
Препарат Эврисди® не оказывает влияния на способность управлять транспортными средствами, механизмами.
Форма выпуска
Порошок для приготовления раствора для приема внутрь 0.75 мг/мл
По 2 г порошка во флакон темного стекла (гидролитический класс III Ф.США/ЕФ) с завинчивающейся пластиковой крышкой, оснащенной уплотнительным диском, с защитой от детей и кольцом первого вскрытия.
1 флакон вместе с размещенными в отдельном прозрачном пакете адаптером из полиэтилена, 2 пероральными шприцами из полипропилена вместимостью 6 мл (помещенными в непрозрачный пакет с маркировкой, обозначающей объем дозирования),
2 пероральными шприцами из полипропилена вместимостью 12 мл (помещенными в непрозрачный пакет с маркировкой, обозначающей объем дозирования), инструкцию по применению и руководство по разведению (только для медицинских работников) помещают в картонную пачку.
С целью контроля первого вскрытия на пачку наносится защитная голографическая наклейка.
Срок годности
2 года.
Не применять по истечении срока годности.
Приготовленный раствор хранить в оригинальном флаконе с плотно закрытой крышкой в вертикальном положении при температуре 2-8 °С в течение 64 дней, не замораживать.
Условия хранения
При температуре не выше 25 °С в оригинальной упаковке (флакон в картонной пачке).
Условия отпуска
Отпускают по рецепту.
Владелец регистрационного удостоверения
Ф. Хоффманн-Ля Рош Лтд., Швейцария
F. Hoffmann-La Roche Ltd, Grenzacherstrasse 124, 4070 Basel, Switzerland
Производитель
Ф. Хоффманн-Ля Рош Лтд., Швейцария
F. Hoffmann-La Roche Ltd, Grenzacherstrasse 124, 4070 Basel, Switzerland
Фасовщик (первичная упаковка)
Ф. Хоффманн-Ля Рош Лтд., Швейцария
F. Hoffmann-La Roche Ltd, Grenzacherstrasse 124, 4070 Basel, Switzerland
Упаковщик (вторичная/потребительская упаковка)
Ф. Хоффманн-Ля Рош Лтд., Швейцария
F. Hoffmann-La Roche Ltd, Wurmisweg, 4303 Kaiseraugst, Switzerland
Выпускающий контроль качества
Ф. Хоффманн-Ля Рош Лтд., Швейцария
F. Hoffmann-La Roche Ltd, Wurmisweg, 4303 Kaiseraugst, Switzerland
Организация, принимающая претензии от потребителей
Претензии потребителей направлять в компанию АО «Рош-Москва» по адресу:
107045, Россия, г. Москва, Трубная площадь, д. 2, помещение 1, этаж 1, комната 42
Руководство по разведению
препарата ЭВРИСДИ® (рисдиплам) в виде раствора для приема внутрь 0.75 мг/мл
Только для медицинских работников
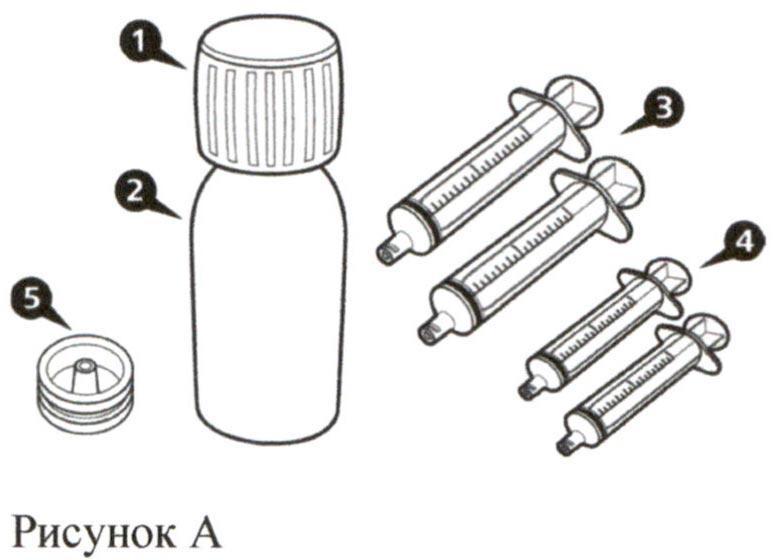
Каждая картонная пачка препарата Эврисди® содержит (см. рисунок А):
- Крышка флакона – 1 шт.
- Флакон с препаратом Эврисди® – 1 шт.
- Пероральные шприцы на 12 мл (в пакетах) – 2 шт.
- Пероральные шприцы на 6 мл (в пакетах) – 2 шт.
- Адаптер для флакона (вдавливаемый) – 1 шт.
- Инструкция по медицинскому применению (не показана на рисунке) – 1 шт.
- Руководство по разведению (не показано на рисунке) – 1 шт.
Важная информация по препарату Эврисди®
- Избегайте вдыхания порошка препарата Эврисди®.
- Используйте перчатки.
- Не используйте порошок, если его срок годности истек. Дата истечения срока годности (Годен до) порошка напечатана на этикетке флакона.
- Не выдавайте приготовленный раствор, если дата утилизации раствора наступит позже, чем дата истечения срока годности исходного порошка.
- Избегайте контакта препарата с Вашей кожей. Если это произошло, промойте данную область водой с мылом.
- Не используйте препарат, если какие-либо элементы упаковки повреждены или отсутствуют.
- Используйте воду очищенную или стерильную воду для инъекций для разведения препарата.
- Не добавляйте другие пероральные шприцы, помимо предоставляемых в картонной пачке.
Как хранить препарат Эврисди®
- Порошок (неразведенный препарат) хранить при комнатной температуре не выше 25 °С в картонной пачке.
- Приготовленный раствор (разведенный препарат) хранить в холодильнике при температуре 2-8 °С.
- Приготовленный раствор для приема внутрь хранить в оригинальном флаконе с плотно закрытой крышкой в вертикальном положении.
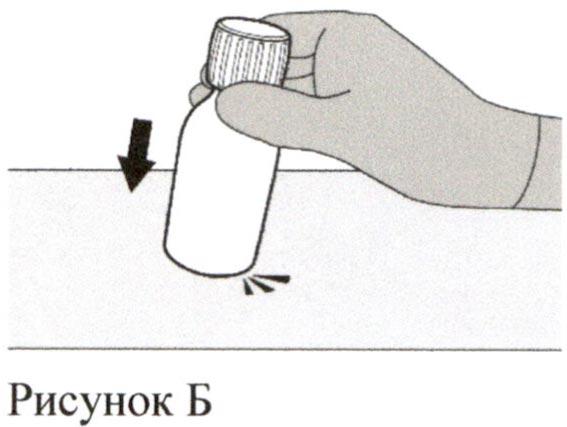
Разведение Шаг 1
Аккуратно постучите по дну флакона для разрыхления порошка (см. Рисунок Б).
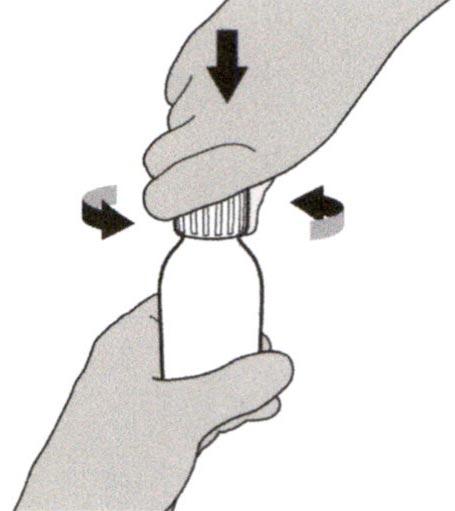
Шаг 2
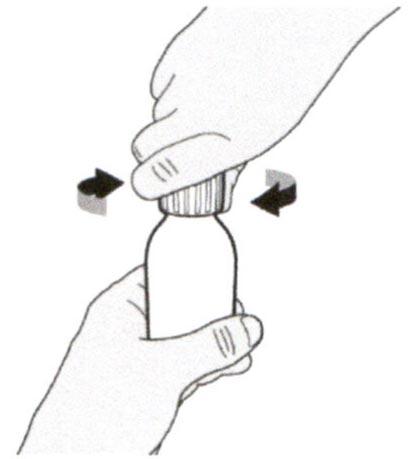
Снимите крышку, нажав на нее вниз и затем повернув налево (против часовой стрелки) (см. Рисунок В). Не выбрасывайте крышку.
Шаг 3
Аккуратно налейте 79 мл воды очищенной или стерильной воды для инъекций во флакон с препаратом (см. Рисунок Г).
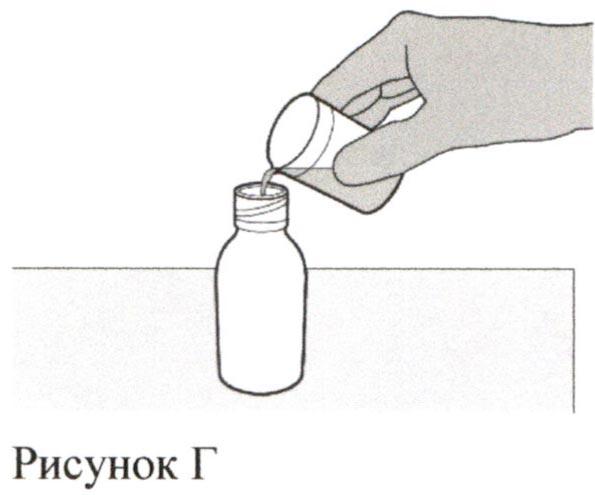
Шаг 4
Удерживайте флакон с препаратом на столе одной рукой.
Вставьте вдавливаемый адаптер для флакона в горлышко флакона, надавливая на адаптер вниз другой рукой. Убедитесь, что адаптер для флакона полностью вдавлен в горлышко флакона (см. Рисунок Д).
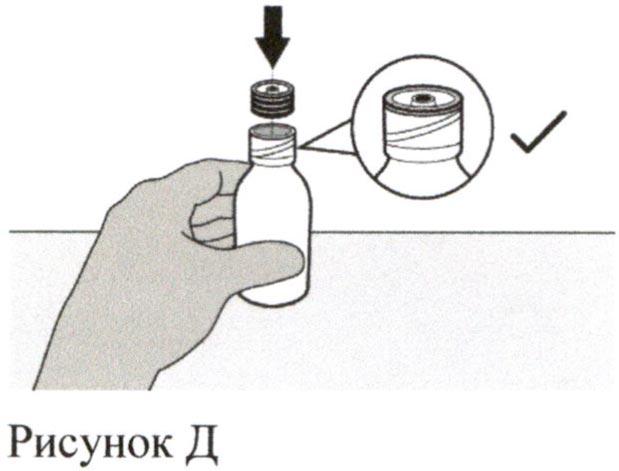
Шаг 5
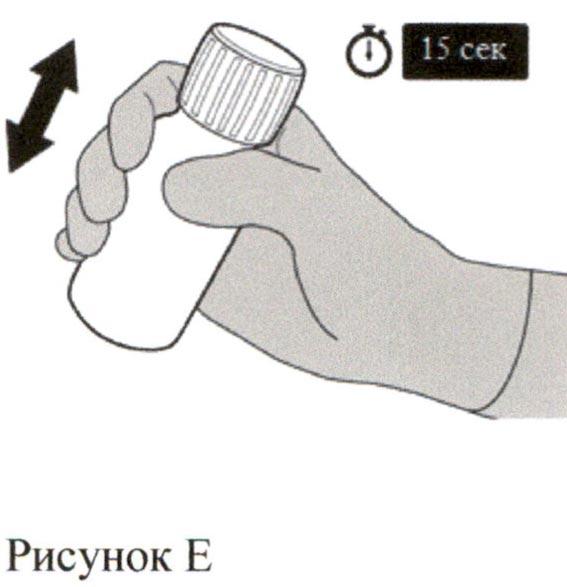
Наденьте крышку обратно на флакон. Поверните крышку вправо (по часовой стрелке), чтобы закрыть флакон.
Убедитесь, что флакон полностью закрыт, и затем хорошо встряхивайте его в течение 15 секунд (см. Рисунок Е).
Подождите 10 минут. У Вас должен получиться прозрачный раствор.
Если этого не произошло, следует хорошо встряхивать флакон в течение 15 секунд еще раз.
Шаг 6
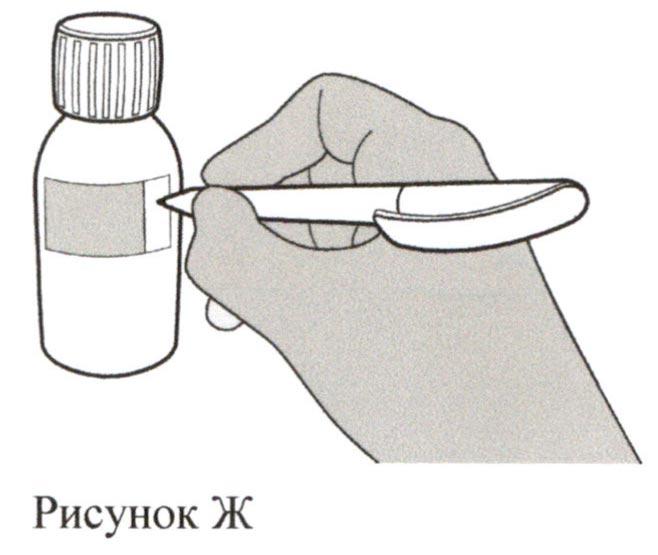
Рассчитайте дату утилизации приготовленного раствора для приема внутрь, равную 64 дням после разведения (Примечание: день разведения считается днем 0. Например, если разведение произведено 1 апреля, датой утилизации будет 4 июня).
Запишите дату утилизации приготовленного раствора на этикетке флакона (см. рисунок Ж) и на картонной пачке.
Поместите флакон обратно в оригинальную картонную пачку вместе со шприцами (в пакетах) и инструкцией по медицинскому применению. Хранить картонную пачку в холодильнике.
Комментарии
ПРАКТИКА ПЕДИАТРА



