Энспринг - инструкция по применению
См. откуда получены инструкции МЕДИ РУ
Регистрационный номер:
ЛП-007335
Общая характеристика лекарственного препарата
▼ Данный лекарственный препарат подлежит дополнительному мониторингу. Это позволит быстро выявить новую информацию по безопасности. Мы обращаемся к работникам системы здравоохранения с просьбой сообщать о любых подозреваемых нежелательных реакциях. Порядок сообщения о нежелательных реакциях представлен в разделе 4.8.
1. Наименование лекарственного препарата
Энспринг, 120 мг/мл, раствор для подкожного введения
2. Качественный и количественный состав
2.1 Общее описание
Сатрализумаб – это рекомбинантное гуманизированное моноклональное антитело к человеческому рецептору интерлейкина-6 (IL-6) из подкласса иммуноглобулинов G2 (IgG2), продуцируемое клетками яичников китайского хомячка по технологии рекомбинантной ДНК (включая технологию рН-зависимого связывания).
2.2 Качественный и количественный состав
Действующее вещество: сатрализумаб.
Каждый мл раствора для подкожного введения содержит 120 мг сатрализумаба.
Каждый шприц-тюбик с 1 мл раствора для подкожного введения содержит 120 мг сатрализумаба.
Полный перечень вспомогательных веществ приведен в разделе 6.1.
3. Лекарственная форма
Раствор для подкожного введения.
Жидкость от прозрачной до сильно опалесцирующей, от бесцветной до слегка желтоватого цвета.
4. Клинические данные
4.1 Показания к применению
Препарат показан к применению у пациентов от 12 лет и старше (12-74 г. согласно КИ).
4.2 Режим дозирования и способ применения
Замена препарата Энспринг на другой препарат биологического происхождения должна быть согласована с лечащим врачом.
Во избежание медицинских ошибок перед применением лекарственного препарата необходимо проверить этикетку на шприц-тюбике и убедиться, что используемый для введения препарат представляет собой препарат Энспринг.
Режим дозирования
Препарат Энспринг можно применять как в виде монотерапии, так и в комбинации с пероральными глюкокортикостероидами, азатиоприном (AZA) или микофенолата мофетилом (ММФ) (см. раздел 5.1).
См. общие характеристики данных лекарственных препаратов.
Нагрузочная доза
Рекомендуемая нагрузочная доза составляет 120 мг подкожно каждые 2 недели (первое введение на неделе 0, второе введение на неделе 2 и третье введение на неделе 4) – всего 3 инъекции.
Поддерживающая доза
Рекомендуемая поддерживающая доза составляет 120 мг подкожно каждые 4 недели.
Продолжительность терапии
Препарат Энспринг предназначен для долгосрочной терапии.
Задержка введения или пропуск дозы
Если инъекция была пропущена, по любой причине за исключением повышения активности ферментов печени, препарат следует ввести как указано в таблице 1.
Таблица 1. Рекомендуемый режим дозирования при задержке введения или пропуске дозы
| Введение последней дозы | Рекомендуемый режим дозирования при задержке введения или пропуске дозы |
| Менее 8 недель в период поддерживающей дозы или при пропуске нагрузочной дозы | Ввести 120 мг путем подкожной инъекции как можно скорее, не дожидаясь следующего запланированного введения. Поддерживающий период При задержке введения или пропуске дозы, скорректировать график применения препарата – каждые 4 недели. Нагрузочный период При задержке введения или пропуске второй нагрузочной дозы следует ввести ее как можно скорее, а затем ввести третью и последнюю нагрузочную дозу через 2 недели. При задержке введения или пропуске третьей нагрузочной дозы следует ввести ее как можно скорее, а затем ввести первую поддерживаю дозу через 4 недели. |
| 8 недель – менее 12 недель | 120 мг путем подкожной инъекции на неделе 0* и 2, затем 120 мг – каждые 4 недели. |
| 12 недель или более | 120 мг путем подкожной инъекции на неделе 0*, 2 и 4, затем 120 мг – каждые 4 недели. |
* Неделя 0 относится к моменту первого введения после пропуска дозы.
Коррекция дозы
Отклонения лабораторных показателей функции печени
При повышении активности аланинаминотрансферазы (АЛТ) или аспартатаминотрансферазы (АСТ) >5х верхней границы нормы (ВГН) с одновременным повышением уровня билирубина (любым) лечение препаратом Энспринг следует прекратить; возобновление лечения не рекомендуется.
При повышении активности АЛТ или АСТ >5х ВГН без одновременного повышения уровня билирубина (любого) терапию препаратом Энспринг следует прекратить; лечение можно возобновить (120 мг подкожно каждые 4 недели) после нормализации активности АЛТ и АСТ на основании оценки пользы и риска для пациента.
В случае, если принято решение о возобновлении терапии препаратом Энспринг, следует тщательно мониторировать показатели функции печени, при любом последующем повышении активности АЛТ/АСТ и/или уровня билирубина следует прекратить применение препарата; возобновление лечения не рекомендуется.
Таблица 2. Рекомендуемый режим дозирования при возобновлении лечения после повышения активности трансаминаз печени
| Введение последней дозы | Рекомендуемый режим дозирования после возобновления лечения |
| Менее 12 недель | Возобновить введение в дозе 120 мг путем подкожной инъекции каждые 4 недели. |
| 12 недель или более | Возобновить введение в дозе 120 мг путем подкожной инъекции на неделе 0*, 2 и 4, затем 120 мг – каждые 4 недели. |
* Неделя 0 относится к моменту первого введения после пропуска дозы.
Нейтропения
Если число нейтрофилов составляет <1.0х109/л и подтверждено повторным тестированием, применение препарата Энспринг следует прервать до тех пор, пока число нейтрофилов не будет >1.0х109/л.
Особые группы пациентов
Пациенты пожилого возраста
Коррекции дозы у пациентов ≥65 лет не требуется (см. разделы 4.4 и 5.2).
Пациенты с нарушением функции почек
Эффективность и безопасность препарата Энспринг у пациентов с нарушением функции почек специально не изучались; однако необходимости в коррекции дозы у пациентов с нарушением функции почек не ожидается (см. разделы 4.4 и 5.2).
Пациенты с нарушением функции печени
Безопасность и эффективность препарата Энспринг у пациентов с нарушением функции печени на данный момент не установлены (см. разделы 4.4 и 5.2).
Дети
Безопасность и эффективность препарата Энспринг у детей <12 лет на данный момент не установлены (см. раздел 4.4). Данные отсутствуют.
Способ применения
Препарат Энспринг следует вводить в виде подкожной инъекции.
Препарат рекомендуется вводить в области живота и бедер. Следует чередовать места инъекций и никогда не вводить препарат в родимые пятна, ткани рубцов, гематомы, в места с уплотнением или повреждением, в участки с чувствительной кожей, покраснением.
Первое введение необходимо осуществлять под наблюдением квалифицированного медицинского специалиста.
По решению лечащего врача взрослый пациент/лицо, осуществляющее уход за пациентом, может вводить препарат Энспринг дома при условии соблюдения техники проведения инъекции.
При появлении симптомов серьезных аллергических реакций пациенты/лица, осуществляющие уход за пациентом, должны незамедлительно обратиться за медицинской помощью и подтвердить с медицинским специалистом возможность продолжения применения препарата Энспринг.
Обращение с препаратом
Шприц-тюбик с препаратом Энспринг предназначен для введения только одной дозы.
При наличии посторонних включений в растворе, его помутнении или изменении окраски не следует вводить препарат.
Необходимо проверить шприц-тюбик на предмет повреждений. При наличии трещин или повреждений не следует вводить препарат.
Препарат Энспринг до применения хранить при температуре 2-8 °С.
Невскрытую картонную пачку (оригинальную упаковку) можно достать и вернуть в холодильник при необходимости. Общее время хранения при комнатной температуре не выше 30 °С (вне холодильника) не должно превышать 8 дней.
Шприц-тюбики необходимо хранить в картонной пачке для защиты от света и всегда сохранять сухими.
Не замораживать. Не использовать, если шприц-тюбик был заморожен.
Не встряхивать.
Использование и утилизация шприц-тюбика
Необходимо строго следовать следующим требованиям по использованию и утилизации шприц-тюбика.
- Шприц-тюбик никогда нельзя использовать повторно.
- Поместить использованный шприц-тюбик в контейнер для утилизации острых предметов сразу после использования.
- Утилизировать шприц-тюбик в соответствии с локальными требованиями или в соответствии с указаниями лечащего врача.
- Хранить шприц-тюбик, как и все лекарственные препараты, в недоступном для детей месте.
4.3 Противопоказания
Гиперчувствительность к сатрализумабу или к любому из вспомогательных веществ, перечисленных в разделе 6.1, в анамнезе.
Одновременное применение с живыми, в том числе ослабленными, вакцинами (см. раздел 4.4).
4.4 Особые указания и меры предосторожности при применении
В медицинской документации пациента следует указывать торговое наименование и номер серии препарата.
С осторожностью
Нарушение функции почек.
Нарушение функции печени.
Одновременный прием или отмена препарата Энспринг у пациентов, получающих субстраты CYP450 с узким терапевтическим индексом.
Язва кишечника или дивертикулит в анамнезе.
Инфекции
Следует отложить введение препарата Энспринг у пациентов с активными инфекциями до взятия активных инфекций под контроль (см. раздел 4.2). Пациентам необходимо предоставить карточку-памятку.
Туберкулез
Туберкулез развивался у пациентов, получающих терапию другими антагонистами рецептора IL-6. Согласно рекомендациям для других иммуномодулирующих средств, перед началом терапии препаратом Энспринг всех пациентов следует проверить на факторы риска, связанные с туберкулезом, и на наличие латентного туберкулеза. Пациентам с активным или латентным туберкулезом в анамнезе следует рассмотреть необходимость проведения антитуберкулезной терапии до начала лечения препаратом Энспринг. Проконсультируйтесь со специалистом по инфекционным заболеваниям, следует ли проводить антитуберкулезную терапию до начала применения препарата Энспринг. Даже если первоначальный тест на туберкулез был отрицательным, пациенты должны находиться под наблюдением на предмет признаков и симптомов туберкулеза во время терапии препаратом Энспринг.
Вакцинация
Не следует одновременно применять препарат Энспринг с живыми, в том числе ослабленными, вакцинами, поскольку клиническая безопасность такого сочетания не установлена (см. раздел 4.3). Интервал между введением живых вакцин и началом терапии препаратом Энспринг определяется в соответствии с действующими руководствами по вакцинации при применении иммуномодулирующих/иммуносупрессивных препаратов.
Данные о влиянии вакцинации на пациентов, получающих препарат Энспринг, отсутствуют. Рекомендуется завершить все мероприятия по иммунизации согласно действующим руководствам по иммунизации перед началом терапии препаратом Энспринг.
Лабораторные показатели функции печени
При применении препарата Энспринг наблюдалось легкое или умеренное повышение активности печеночных трансаминаз. Большинство случаев повышений было <5х ВГН, не приводили к приостановке или прекращению лечения и разрешались в период терапии препаратом Энспринг.
Следует мониторировать активность АЛТ и АСТ каждые 4 недели в течение первых трех месяцев терапии, затем каждые 3 месяца в течение одного года, далее – в зависимости от клинических показаний. Рекомендации по прекращению терапии см. в разделе 4.2, подраздел «Коррекция дозы».
Реактивация гепатита В
При применении ИСТ наблюдались случаи реактивации гепатита В. Пациенты с положительными результатами анализов на гепатит исключались из клинических исследований препарата Энспринг. Перед началом терапии препаратом Энспринг всем пациентам следует провести скрининг на вирус гепатита B в соответствии с местными стандартами. Пациентов с активным гепатитом В (активная инфекция, подтвержденная положительными результатами теста на поверхностный антиген вируса гепатита В (HBsAg) и положительными результатами теста на антитела к вирусу гепатита B (anti-HBV), не следует лечить препаратом Энспринг. Пациенты с отрицательным результатом по HBsAg и положительные по антителам к коровому антигену вируса гепатита В (HB core antibody, HBcAb+)) или с положительными результатами теста на носительство гепатита B (HBsAg+) должны проконсультироваться с гепатологом перед началом лечения и наблюдаться в соответствии с местными стандартами с целью предотвращения реактивации гепатита B.
Число нейтрофилов
При применении препарата Энспринг наблюдалось снижение числа нейтрофилов (см. раздел 4.8).
Следует мониторировать число нейтрофилов через 4-8 недель после начала терапии, далее – в зависимости от клинических показаний. Рекомендации по прерыванию терапии см. в разделе 4.2, подраздел «Коррекция дозы».
Злоупотребление приемом препарата или лекарственная зависимость
Исследования по изучению злоупотребления приемом препарата или лекарственной зависимости не проводились. Однако, согласно доступным данным нет доказательств того, что применение препарата Энспринг может привести к лекарственной зависимости.
Злокачественные новообразования
Иммуномодулирующие средства могут повышать риск развития злокачественных новообразований. Влияние лечения препаратом Энспринг на развитие злокачественных новообразований неизвестно.
Активация системы комплемента
Терапия ингибиторами рецепторов IL-6 может привести к активации системы комплемента. На основании имеющихся клинических данных при применении препарата Энспринг такого влияния не наблюдалось. Однако, в связи с ограниченными данными, этот риск при применении препарата Энспринг не может быть точно оценен.
Демиелинизирующие заболевания центральной нервной системы (ЦНС)
В ходе терапии ингибиторами рецепторов IL-6 отмечались случаи развития других воспалительных заболеваний ЦНС. Следует обратить внимание на симптомы, указывающие на развитие демиелинизирующего заболевания ЦНС. Однако, в связи с ограниченными данными, этот риск при применении препарата Энспринг не может быть точно оценен.
Перфорация дивертикула/кишечника
В ходе терапии ингибиторами рецепторов IL-6 у пациентов с ревматоидным артритом отмечались случаи перфорации дивертикула и кишечника. Пациенты с дивертикулитом в анамнезе исключались из опорных исследований препарата Энспринг. Повышенный риск перфорации дивертикула/кишечника, который отмечался при применении других ингибиторов рецепторов IL-6, не может быть исключен при лечении препаратом Энспринг. У пациентов с язвой кишечника или дивертикулитом в анамнезе препарат Энспринг следует применять с осторожностью. В случае острой боли в животе пациентов следует незамедлительно проверить для раннего обнаружения перфорации органов желудочно-кишечного тракта.
Особые группы пациентов
Пациенты пожилого возраста
Безопасность и эффективность препарата Энспринг изучались у пациентов ≤74 лет (см. разделы 4.2 и 5.2).
Безопасность и эффективность препарата Энспринг у пациентов >74 лет не изучались (см. раздел 4.2).
Пациенты с нарушением функции почек
Безопасность и эффективность препарата Энспринг у пациентов с нарушением функции почек специально не изучались, однако препарат Энспринг представляет собой моноклональное антитело и преимущественно выводится путем катаболизма (а не почками). Таким образом, не ожидается, что пациентам с нарушением функции почек потребуется коррекция дозы. Пациенты с нарушением функции почек легкой степени тяжести принимали участие в клинических исследованиях, фармакокинетика сатрализумаба у таких пациентов не изменялась (см. разделы 4.2 и 5.2).
Пациенты с нарушением функции печени
Безопасность и эффективность препарата Энспринг у пациентов с нарушением функции печени не изучались (см. разделы 4.2 и 5.2).
Дети
Безопасность и эффективность препарата Энспринг изучали у ограниченного числа подростков ≥12 лет. Результаты изучения фармакокинетики, эффективности и безопасности соответствовали таковым у взрослых (см. разделы 5.1 и 5.2).
Безопасность и эффективность препарата Энспринг у пациентов <12 лет еще не изучались (см. раздел 4.2).
4.5 Взаимодействие с другими лекарственными препаратами и другие виды взаимодействия
Отдельных исследований лекарственных взаимодействий для препарата Энспринг не проводилось.
Популяционный фармакокинетический анализ не выявил какого-либо влияния AZA, глюкокортикостероидов или ММФ на клиренс препарата Энспринг.
Возможное влияние препарата Энспринг на снижение экспозиции одновременно применяемых препаратов, метаболизирующихся изоферментами цитохрома CYP450, посредством блокады сигнального пути IL-6, изучали с использованием различных подходов на фармакокинетической модели, основанной на физиологии.
Было показано, что подавление передачи сигнала IL-6 при терапии препаратом Энспринг от низких исходных уровней IL-6, отмечавшихся в клинических исследованиях фазы III, будет оказывать только небольшое влияние на экспозицию диапазона проб субстратов CYP450 (≤15% уменьшение площади под кривой «концентрация-время» (AUC) для всех субстратов CYP 1A2, 3A4, 2D6, 2C19). Это указывает на то, что риск лекарственного взаимодействия является низким, однако при применении препарата Энспринг или его отмене у пациентов, одновременно получающих субстраты CYP450 с узким терапевтическим индексом, следует соблюдать осторожность.
Пациенты, принимающие индивидуально контролируемые дозы препаратов, метаболизируемых CYP450 3А4, 1А2 или 2С9 (например, аторвастатин, блокаторы кальциевых каналов, теофиллин, варфарин, фенитоин, циклоспорин или бензодиазепины), должны находиться под наблюдением в момент начала и в момент окончания терапии препаратом Энспринг, при необходимости доза этих веществ может быть скорректирована. Учитывая длительный период полувыведения, влияние препарата Энспринг на активность ферментов CYP450 может сохраняться в течение нескольких недель после прекращения лечения.
4.6 Фертильность, беременность и лактация
Беременность
Данные о применении сатрализумаба у беременных женщин отсутствуют. Исследования репродуктивной токсичности на животных не свидетельствуют о наличии репродуктивной токсичности (см. раздел 5.3).
Применение препарата Энспринг беременными женщинами противопоказано.
Лактация
Неизвестно, выводится ли препарат Энспринг с грудным молоком у человека или подвергается системной абсорбции после проглатывания с молоком. Тем не менее, поскольку IgG выводятся с грудным молоком у человека, и существуют данные о выведении сатрализумаба с грудным молоком у животных (см. раздел 5.3), применение препарата Энспринг в период грудного вскармливания противопоказано.
Фертильность
Данные клинических исследований по влиянию препарата Энспринг на фертильность у человека отсутствуют. Результаты изучения препарата у животных показали отсутствие влияния на фертильность самцов и самок (см. раздел 5.3).
4.7 Влияние на способность управлять транспортными средствами и работать с механизмами
Исследований по изучению влияния препарата Энспринг на способность управлять транспортными средствами и механизмами не проводилось. Однако, согласно доступным данным нет доказательств того, что применение препарата Энспринг может оказывать влияние на способность управлять транспортными средствами и механизмами.
4.8 Нежелательные реакции
Резюме профиля безопасности
Безопасность лечения препаратом Энспринг изучали в двух опорных клинических исследованиях, в которых 63 пациента получали препарат Энспринг в виде монотерапии, а 41 – препарат Энспринг в комбинации с иммуносупрессивной терапией. Медиана экспозиции к сатрализумабу составила приблизительно 2 года в обоих исследованиях (в каждом).
Наиболее частыми нежелательными реакциями были головная боль, артралгия, лейкопения и реакции, связанные с инъекцией.
Помимо указанных в разделе нежелательных реакций возможны также перфорация кишечника, оппортунистические инфекции, включая туберкулез, реактивация гепатита В, повышенный риск сердечно-сосудистых заболеваний, активация системы комплемента. Так же не могут быть исключены демиелинизирующие заболевания и злокачественные новообразования (см. раздел 4.4).
Ниже представлены нежелательные реакции, которые были связаны с применением препарата Энспринг в виде монотерапии или в комбинации с иммуносупрессивной терапией в клинических исследованиях. В данном разделе нежелательные реакции сгруппированы в соответствии с классами систем органов медицинского словаря для нормативно-правовой деятельности MedDRA.
Для описания частоты нежелательных реакций используется следующая классификация: очень часто (≥1/10), часто (≥1/100, но <1/10), нечасто (≥1/1000, но <1/100), редко (≥1/10000, но <1/1000), очень редко (<1/10000).
Нарушения со стороны крови и лимфатической системы: часто – гипофибриногенемия.
Психические нарушения: часто – бессонница.
Нарушения со стороны нервной системы: очень часто – головная боль; часто – мигрень.
Нарушения со стороны дыхательной системы, органов грудной клетки и средостения: часто – аллергический ринит.
Нарушения со стороны кожи и подкожных тканей: часто – сыпь, зуд.
Нарушения со стороны скелетно-мышечной и соединительной ткани: очень часто – артралгия; часто – скелетно-мышечная скованность.
Общие расстройства и нарушения в месте введения: часто – периферический отек.
Травмы, интоксикации и осложнения процедур: очень часто – реакции, связанные с инъекцией.
Лабораторные и инструментальные данные: очень часто – снижение числа лейкоцитов; часто – повышение уровня билирубина.
Описание отдельных нежелательных реакций
Реакции, связанные с инъекцией
Реакции, связанные с инъекцией, наблюдались у пациентов, получающих препарат Энспринг как в виде монотерапии, так и в комбинации с ИСТ; были преимущественно от легкой до умеренной степени тяжести, большинство развивались в течение 24 часов после инъекции. Наиболее частыми системными симптомами были диарея и головная боль. Наиболее частыми локальными реакциями, связанными с инъекцией, были гиперемия, эритема, зуд, сыпь и боль. Ни одна реакция, связанная с инъекцией, не привела к необходимости прервать введение или прекратить применение препарата.
Инфекции
В исследовании монотерапии препаратом Энспринг частота инфекций была ниже у пациентов, получающих препарат (99.8 явлений/100 пациенто-лет (95% доверительный интервал (ДИ): 82.4, 119.8) по сравнению с пациентами, получающими плацебо (162.6 явлений/100 пациенто-лет (95% ДИ: 125.8, 206.9)). Частота серьезных инфекций составила 5.2 явлений/100 пациенто-лет (95% ДИ: 1.9, 11.3) у пациентов, получающих препарат Энспринг, по сравнению с 9.9 явлений/100 пациенто-лет (95% ДИ: 2.7, 25.2) у пациентов, получающих плацебо.
У пациентов, получающих препарат Энспринг в комбинации с ИСТ, частота инфекций составила 132.5 явлений/100 пациенто-лет (95% ДИ: 108.2, 160.5) по сравнению с 149.6 явлений/100 пациенто-лет (95% ДИ: 120.1, 184.1) у пациентов, получающих плацебо в комбинации с ИСТ. У пациентов, получающих препарат Энспринг в комбинации с ИСТ, частота серьезных инфекций составила 2.6 явлений/100 пациенто-лет (95% ДИ: 0.3, 9.2) по сравнению с 5.0 явлений/100 пациенто-лет (95% ДИ: 1.0, 14.7) у пациентов, получающих плацебо в комбинации с ИСТ.
Увеличение массы тела
В клинических исследованиях препарата Энспринг® в качестве монотерапии или в комбинации с ИСТ увеличение массы тела ≥15% от исходного уровня наблюдалось у 3.8% пациентов по сравнению с 2.7% пациентов, получавших плацебо (или в комбинации с ИСТ).
Отклонения со стороны лабораторных показателей
Нейтрофилы
В период лечения снижение показателя нейтрофилов наблюдалось у 31.7% пациентов, получающих препарат Энспринг (в виде монотерапии или в комбинации с ИСТ), по сравнению с 21.6% пациентов, получающих плацебо (или плацебо в комбинации с ИСТ). Большинство случаев снижения нейтрофилов было временным или периодическим.
У 9.6% пациентов в группе терапии препаратом Энспринг показатель нейтрофилов составил <1х109/л по сравнению с 5.4% в группе пациентов, получающих плацебо или плацебо в комбинации с ИСТ, что не было связано по времени с какими-либо серьезными инфекциями.
Тромбоциты
В период лечения снижение числа тромбоцитов наблюдалось у 24.0% пациентов, получающих препарат Энспринг (в виде монотерапии или в комбинации с ИСТ), по сравнению с 9.5% пациентов, получающих плацебо или плацебо в комбинации с ИСТ. Снижение числа тромбоцитов не было связано с явлениями кровотечений.
Большинство случаев снижения числа тромбоцитов было временными, число тромбоцитов при этом было не ниже 75х109/л. Ни у одного из пациентов не отмечалось снижения числа тромбоцитов ≤50 х109/л.
Показатели функции печени
В период лечения повышение активности АЛТ или АСТ наблюдалось у 27.9% и 18.3% пациентов, получающих препарат Энспринг (в виде монотерапии или в комбинации с ИСТ), соответственно, по сравнению с 12.2% и 13.5% пациентов, получающих плацебо или плацебо в комбинации с ИСТ. В большинстве случаев повышение активности АЛТ или АСТ было ниже 3х ВГН, временным и разрешалось без прерывания терапии препаратом Энспринг.
Повышение активности АЛТ или АСТ >3х ВГН, которое не было связано с повышением показателя общего билирубина, отмечалось у 2.9% и 1.9% пациентов, получающих препарат Энспринг в виде монотерапии или в комбинации с ИСТ, соответственно. Повышение активности АЛТ >5х ВГН отмечалось у одного пациента через 4 недели после начала терапии препаратом Энспринг в комбинации с ИСТ. Показатель АЛТ нормализовался после отмены препарата Энспринг.
Липидный профиль
В период лечения повышение показателя общего холестерина >7.75 ммоль/л наблюдалось у 10.6% пациентов, получающих препарат Энспринг (в виде монотерапии или в комбинации с ИСТ), по сравнению с 1.4% пациентов, получающих плацебо или плацебо в комбинации с ИСТ; у 20.2% пациентов, получающих препарат Энспринг, отмечалось повышение показателя триглицеридов >3.42 ммоль/л по сравнению с 10.8% пациентов, получающих плацебо. Повышение показателей липидного профиля не потребовало прерывания терапии препаратом Энспринг.
Сообщение о подозреваемых нежелательных реакциях
Важно сообщать о подозреваемых нежелательных реакциях после регистрации лекарственного препарата с целью обеспечения непрерывного мониторинга соотношения «польза-риск» лекарственного препарата. Медицинским работникам рекомендуется сообщать о любых подозреваемых нежелательных реакциях лекарственного препарата через национальные системы сообщения о нежелательных реакциях государств – членов Евразийского экономического союза.
Республика Армения
АОЗТ «Научного Центра Экспертизы Лекарств и Медицинских Технологий им. Академика Э. Габриеляна»
0051, г. Ереван, пр. Комитаса, д. 49/4
www.pharm.am
Республика Беларусь
Республиканское унитарное предприятие «Центр экспертиз и испытаний в здравоохранении»
220037, г. Минск, пер. Товарищеский, д. 2а
www.rceth.by
Кыргызская Республика
Департамент лекарственных средств и медицинских изделий при Министерстве здравоохранения Кыргызской Республики
720044, г. Бишкек, ул. 3-я Линия, д. 25
www.pharm.kg
Российская Федерация
Федеральная служба по надзору в сфере здравоохранения (Росздравнадзор)
109012, г. Москва, Славянская площадь, д. 4, стр. 1
www.roszdravnadzor.gov.ru
4.9 Передозировка
Случаев передозировки препарата у пациентов с оптиконевромиелитом и заболеваниями спектра оптиконевромиелита не было.
Симптомы
В клиническом исследовании фазы I препарат Энспринг вводили здоровым взрослым добровольцам однократно в дозе ≤240 мг подкожно; серьезных или тяжелых нежелательных явлений не отмечалось.
Лечение
В случае передозировки следует тщательно наблюдать за пациентом, и при необходимости провести симптоматическую или поддерживающую терапию.
5. Фармакологические свойства
5.1 Фармакодинамические свойства
Фармакотерапевтическая группа: Иммунодепрессанты; ингибиторы интерлейкинов.
Код АТХ: L04AC19
В клинических исследованиях препарата Энспринг у пациентов с оптиконевромиелитом и заболеваниями спектра оптиконевромиелита отмечалось снижение показателей C-реактивного белка, фибриногена и комплемента (C3, C4 и CH50).
Механизм действия
Сатрализумаб представляет собой гуманизированное моноклональное антитело из подкласса иммуноглобулинов G2 (IgG2), которое связывается как с растворимым, так и с мембранным человеческим рецептором интерлейкина-6 (IL-6R), и таким образом предотвращает передачу сигнала по нисходящему пути через эти рецепторы.
IL-6 является многофункциональным цитокином, который продуцируется различными типами клеток и участвует в различных воспалительных процессах, таких как активация В-клеток, дифференцировка В-клеток в плазмобласты и выработка аутоантител, Th17-клеточная активация и дифференцировка, ингибирование регуляторных Т-клеток, изменение проницаемости гематоэнцефалического барьера. Уровни IL-6 повышаются у пациентов с оптиконевромиелитом и заболеваниями спектра оптиконевромиелита в спинномозговой жидкости и сыворотке крови в периоды активности заболевания. Некоторые функции IL-6 вовлечены в патогенез оптиконевромиелита и заболеваний спектра оптиконевромиелита, включая выработку патологических аутоантител к AQP4 (аквапорин-4, белок семейства водных каналов, который, главным образом, экспрессируется астроцитами в ЦНС).
Клиническая эффективность и безопасность
Эффективность и безопасность препарата Энспринг оценивали в двух опорных клинических исследованиях фазы III (BN40898 и BN40900) у пациентов с оптиконевромиелитом с антителами к аквапорину-4 (AQP4-IgG) или без них (по критериям Вингерчука, 2006 г.) и у пациентов с заболеваниями спектра оптиконевромиелита с AQP4-IgG или без них (по критериям Вингерчука, 2007 г.). Ретроспективно можно сказать, что пациенты также соответствовали последним критериям, предложенным международной комиссией по диагнозу оптиконевромиелита (критерии Вингерчука и др., 2015). Действие препарата Энспринг изучали у взрослых пациентов (в исследованиях BN40898 и BN40900) и у подростков (от 12 до 18 лет) (в исследовании BN40898). Доля взрослых пациентов с оптиконевромиелитом без AQP4-IgG, включенных в оба исследования составляла приблизительно 30%, что отражает реальную популяцию пациентов с оптиконевромиелитом.
Первичным показателем эффективности в обоих исследованиях было возникновение рецидивов в соответствии с протоколом на основании заранее определенного ухудшения по расширенной шкале оценки степени инвалидизации (EDSS) и оценок функциональных систем (FSS) с подтверждением независимым комитетом по оценке клинических конечных точек (CEC, Clinical Endpoint Committee). В анализе первичной конечной точки рассматривали время до возникновения первого, подтвержденного CEC рецидива в соответствии с протоколом, с оценкой по шкалам EDSS/FSS в течение 7 дней после того, как пациент сообщил о симптомах (признанный рецидив).
Исследование BN40898 (также известное как SA-307JG или SAkuraSky)
Исследование BN40898 представляло собой рандомизированное, многоцентровое, двойное слепое, плацебо-контролируемое клиническое исследование по оценке действия препарата Энспринг при применении в комбинации с базисной ИСТ (пероральные глюкокортикостероиды в дозе до 15 мг/сутки [аналог преднизолона], AZA в дозе до 3 мг/кг/сутки или ММФ в дозе до 3000 мг/сутки; подростки получали комбинацию AZA и пероральных глюкокортикостероидов или ММФ и пероральные глюкокортикостероиды). В исследование было включено 83 пациента с AQP4-IgG и без них (включая 7 подростков). Пациенты получали 3 первые дозы препарата Энспринг 120 мг или соответствующее плацебо путем подкожных инъекций в область живота или бедра один раз в 2 недели в течение первых 4 недель и один раз в 4 недели в дальнейшем.
Дизайн исследования и исходные характеристики исследуемой популяции представлены в таблице 3.
Исследование определялось на основе наступления событий, и двойной слепой период оценки эффективности был завершен после регистрации 26 признанных случаев рецидива. Пациенты, у которых возник рецидив, подтвержденный CEC, в соответствии с протоколом или пациенты, которые получали неотложную терапию по поводу рецидива в течение двойного слепого периода или пациенты, которые завершили двойной слепой период, могли быть переведены в расширенный открытый период, в котором все пациенты получали открытое лечение препаратом Энспринг.
Таблица 3. Дизайн и исходные характеристики исследования BN40898
| Название исследования | Исследование BN40898 (N=83) | ||
| Дизайн исследования | |||
| Исследуемая популяция | Подростки и взрослые пациенты с оптиконевромиелитом или заболеваниями спектра оптиконевромиелита, получавшие базисную ИСТ. Возраст от 12 до 74 лет, ≥2 рецидивов за последние 2 года, предшествовавшие скринингу (по меньшей мере один рецидив в течение 12 месяцев, предшествовавших скринингу), оценка по шкале EDSS от 0 до 6.5 балла. | ||
| Продолжительность исследования для оценки эффективности | Определяется на основе наступления событий (26 подтвержденных CECрецидивов в соответствии с протоколом). Медиана периода последующего наблюдения: Энспринг – 115.1 недели, плацебо – 42.5 недели | ||
| Группы лечения при рандомизации в соотношении 1:1 | Группа А: Энспринг, 120 мг подкожно Группа B: плацебо | ||
| Исходные характеристики | Энспринг + ИСТ (n=41) | Плацебо + ИСТ (n=42) | |
| Диагноз, n (%): | оптиконевромиелит | 33 (80.5) | 28 (66.7) |
| заболевание спектра оптиконевромиелита | 8 (19.5) | 14 (33.3) | |
| Серопозитивный статус AQP4-IgG, n (%) | 27 (65.9) | 28 (66.7) | |
| Средний возраст в годах (СО), (мин.- макс.) | 40.8 (16.1) (13-73) | 43.4 (12.0) (14-65) | |
| Подростки (в возрасте от ≥12 до <18 лет), n (%) | 4 (9.8) | 3 (7.1) | |
| Распределение по полу, n (%) мужской пол / n (%) женский пол | 4 (9.8) / 37 (90.2) | 2 (4.8) / 40 (95.2) | |
| ИСТ, n (%): | Пероральные глюкокортикостероиды (ПКС) | 17 (41.5) | 20 (47.6) |
| AZA | 16 (39.0) | 13 (31.0) | |
| ММФ | 4 (9.8) | 8 (19.0) | |
| AZA + ПКС* | 3 (7.3) | 0 | |
| ММФ + ПКС* | 1 (2.4) | 1 (2.4) | |
* Комбинация, разрешенная для применения у подростков.
Исследование BN40900 (также известное как SA-309JG или SAkuraStar)
Исследование BN40900 представляло собой рандомизированное, многоцентровое, двойное слепое, плацебо-контролируемое клиническое исследование по оценке монотерапии препаратом Энспринг в сравнении с плацебо. В исследование было включено 95 взрослых пациентов с AQP4-IgG и без них. Пациенты получали 3 первые дозы лекарственного препарата Энспринг 120 мг или соответствующее плацебо путем подкожной инъекции в область живота или бедра один раз в 2 недели в течение первых 4 недель и один раз в 4 недели в дальнейшем.
Дизайн исследования и исходные характеристики исследуемой популяции представлены в таблице 4.
Двойной слепой период оценки эффективности был завершен через 1.5 года после даты рандомизации последнего включенного в исследование пациента. Пациенты, у которых возник подтвержденный CEC рецидив в соответствии с протоколом или пациенты, которые завершили двойной слепой период, могли быть переведены в расширенный открытый период, в котором все пациенты получали открытое лечение препаратом Энспринг.
Таблица 4. Дизайн исследования и исходные характеристики для исследования BN40900
| Название исследования | Исследование BN40900 (N=95) | ||
| Дизайн исследования | |||
| Исследуемая популяция | Взрослые пациенты с оптиконевромиелитом или заболеваниями спектра оптиконевромиелита Возраст от 18 до 74 лет, ≥1 рецидива или дебют заболевания в течение последних 12 месяцев, предшествовавших скринингу, оценка по шкале EDSS от 0 до 6.5 балла. Пациенты либо получали ранее терапию для предотвращения рецидивов заболеваний спектра оптиконевромиелита, либо ранее не получали терапию. | ||
| Продолжительность исследования для оценки эффективности | Определяется на основе наступления событий (44 подтвержденных CEC случаев рецидива в соответствии с протоколом либо 1.5 года с даты рандомизации последнего включенного в исследование пациента, в зависимости от того, какое событие наступит раньше) Медиана периода последующего наблюдения: Энспринг – 95.4 недели, плацебо – 60.5 недели | ||
| Группы лечения при рандомизации в соотношении 2:1 | Монотерапия: Группа А: Энспринг, 120 мг подкожно Группа B: плацебо | ||
| Исходные характеристики | Энспринг (n = 63) | Плацебо (n = 32) | |
| Диагноз, n (%): | оптиконевромиелит | 47 (74.6) | 24 (75.0) |
| заболевания спектра оптиконевромиелита | 16 (25.4) | 8 (25.0) | |
| Серопозитивный статус AQP4-IgG, n (%) | 41 (65.1) | 23 (71.9) | |
| Средний возраст в годах (СО) (мин.-макс.) | 45.3 (12.0) (21-70) | 40.5 (10.5) (20-56) | |
| Распределение по полу, n (%) мужской пол / n (%) женский пол | 17 (27.0) / 46 (73.0) | 1 (3.1) / 31 (96.9) | |
Первичная оценка эффективности – двойной слепой период
Лечение препаратом Энспринг привело к статистически значимому снижению риска возникновения признанного рецидива на 62% (отношение рисков [ОР] [95% ДИ]: 0.38 [0.16-0.88]; p [лог-ранговый критерий] = 0.0184) при применении в комбинации с базисной ИСТ (исследование BN40898) и к снижению риска возникновения признанного рецидива на 55% (ОР [95% ДИ]: 0.45 [0.23-0.89]; p = 0.0184) при применении в виде монотерапии (исследование BN40900) в сравнении с плацебо. На 48 неделе у 88.9% и 76.1% пациентов, получавших препарат Энспринг, по-прежнему не было зарегистрировано признанного рецидива при применении исследуемого препарата в комбинации с ИСТ или в виде монотерапии, соответственно. На 96 неделе у 77.6% и 72.1% пациентов, получавших препарат Энспринг по-прежнему не было зарегистрировано признанного рецидива при применении исследуемого препарата в комбинации с ИСТ или в виде монотерапии, соответственно. После объединения данных двух исследований было отмечено, что терапия препаратом Энспринг приводила к снижению риска развития признанного рецидива на 58% в сравнении с плацебо (ОР [95% ДИ]: 0.42 [0.25-0.71]; p = 0.0008) (см. таблицу 5, рисунки 1 и 2).
Наибольший эффект в подгруппе наблюдался среди пациентов с AQP4-IgG. У таких пациентов относительный риск возникновения признанного рецидива снижался на 79% (ОР [95% ДИ]: 0.21 [0.06-0.75]) в исследовании BN40898 и на 74% (ОР [95% ДИ]: 0.26 [0.11-0.63]) в исследовании BN40900. На 48 неделе у 91.5% и 82.9% пациентов с AQP4-IgG, получавших препарат Энспринг, по-прежнему не было зарегистрировано признанного рецидива при применении исследуемого препарата в комбинации с ИСТ или в виде монотерапии соответственно. На 96 неделе у 91.5% и 76.5% пациентов с AQP4-IgG, получавших препарат Энспринг по-прежнему не было зарегистрировано признанного рецидива при применении исследуемого препарата в комбинации с ИСТ или в виде монотерапии соответственно. При объединении данных, полученных в исследованиях BN40898 и BN40900, было отмечено, что лечение препаратом Энспринг в виде монотерапии или в комбинации с ИСТ привело к общему снижению риска на 75% (ОР [95% ДИ]: 0,25 [0.12-0.50]) у пациентов с AQP4-IgG (см. таблицу 5, рисунки 3 и 4). Значимые различия во времени до возникновения первого признанного рецидива у пациентов без AQP4-IgG между группой, получавшей препарат Энспринг в виде монотерапии или в комбинации с ИСТ, и группой, получавшей плацебо в виде монотерапии или в комбинации с ИСТ, не наблюдались (объединенные данные исследований BN40898 и BN40900: ОР [95% ДИ]: 0.97 [0.41-2.33]).
Таблица 5. Ключевые конечные точки оценки эффективности в исследованиях BN40898 и BN40900
| BN40898 | BN40900 | |||
| Энспринг + ИСТ | Плацебо + ИСТ | Энспринг | Плацебо | |
| (n = 41) | (n = 42) | (n = 63) | (n = 32) | |
| Первичная конечная точка | ||||
| Снижение риска (отдельные исследования) | 62% (ОР: 0.38; 95% ДИ: 0.16; 0.88; p = 0.0184) | 55% (ОР: 0.45; 95% ДИ: 0.23; 0.89; p = 0.0184) | ||
| Снижение риска (объединенный анализ) | 58% (ОР: 0.42 ; 95% ДИ: 0.25; 0.71; p = 0.0008) | |||
| Доля пациентов без признанного рецидива на 48 неделе | 88.9% (95% ДИ: 72.81; 95.70) | 66.0% (95% ДИ: 47.65; 79.25) | 76.1% (95% ДИ: 63.55; 84.86) | 61.9% (95% ДИ: 42.66; 76.26) |
| Доля пациентов без признанного рецидива на 96 неделе | 77.6% (95% ДИ: 58.08; 88.82) | 58.7% (95% ДИ: 39.85; 73.43) | 72.1% (95% ДИ: 58.91; 81.75) | 51.2% (95% ДИ: 32.36; 67.23) |
| Анализ первичной конечной точки в подгруппах (пациенты с AQP4-IgG) | ||||
| Количество пациентов с AQP4-IgG (n) | 27 | 28 | 41 | 23 |
| Снижение риска (отдельные исследования) | 79% (ОР: 0.21; 95% ДИ: 0.06; 0.75; p = 0.0086) | 74% (ОР: 0.26; 95% ДИ: 0.11; 0.63; p = 0.0014) | ||
| Снижение риска (объединенный анализ) | 75% (ОР: 0.25; 95% ДИ: 0.12; 0.50; p: <0.0001) | |||
| Доля пациентов без признанного рецидива на 48 неделе | 91.5% (95% ДИ: 69.64; 97.83) | 59.9% (95% ДИ: 36.25; 77.25) | 82.9% (95% ДИ: 67.49; 91.47) | 55.4% (95% ДИ: 32.96; 73.08) |
| Доля пациентов без признанного рецидива на 96 неделе | 91.5% (95% ДИ: 69.64; 97.83) | 53.3% (95% ДИ: 29.34; 72.38) | 76.5% (95% ДИ: 59.22; 87.21) | 41.1% (95% ДИ: 20.76; 60.41) |
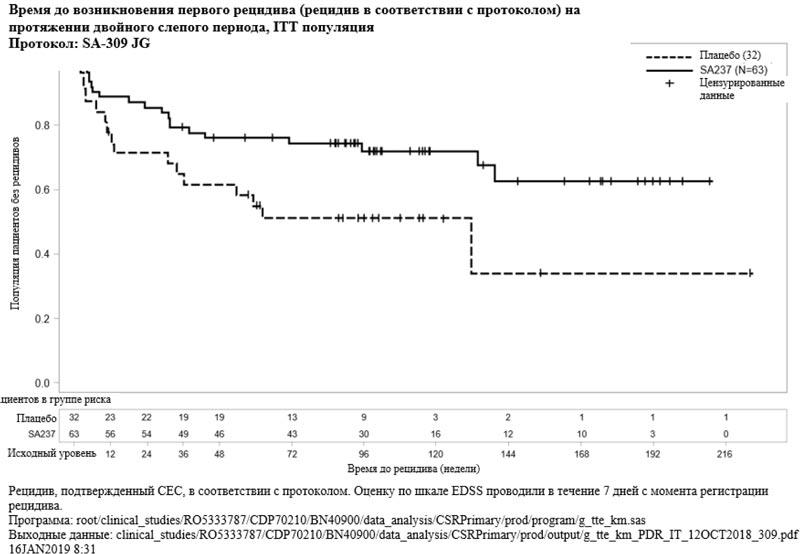 Рисунок 1. Исследование BN40898: время до возникновения первого признанного рецидива (ITT популяция)
Рисунок 1. Исследование BN40898: время до возникновения первого признанного рецидива (ITT популяция)
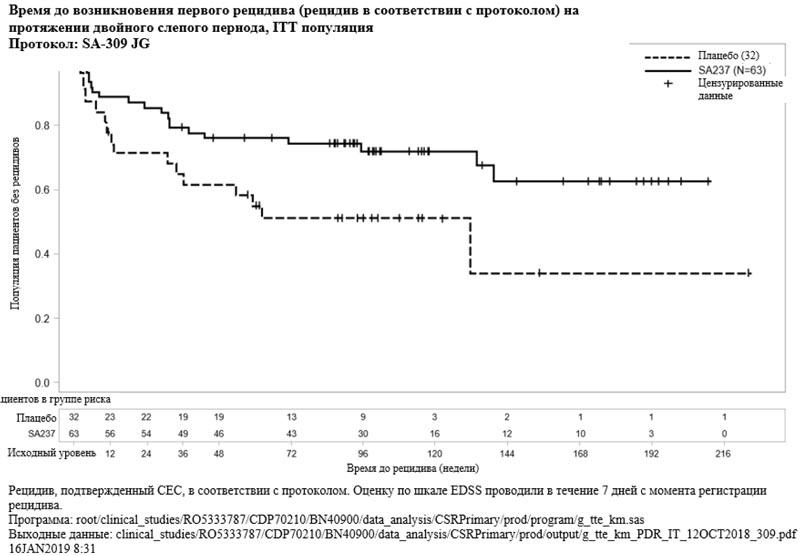 Рисунок 2. Исследование BN40900: время до возникновения первого признанного рецидива в течение двойного слепого периода (ITT популяция)
Рисунок 2. Исследование BN40900: время до возникновения первого признанного рецидива в течение двойного слепого периода (ITT популяция) 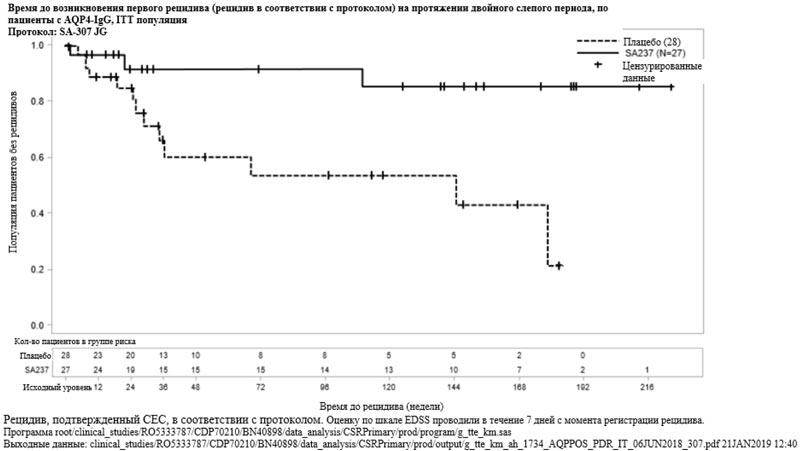 Рисунок 3. Исследование BN40898: время до возникновения первого признанного рецидива в течение двойного слепого периода у пациентов с AQP4-IgG
Рисунок 3. Исследование BN40898: время до возникновения первого признанного рецидива в течение двойного слепого периода у пациентов с AQP4-IgG 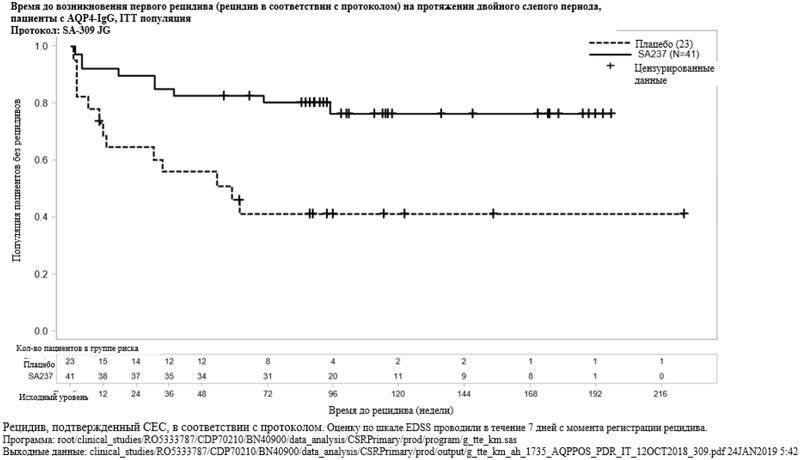 Рисунок 4. Исследование BN40900: время до возникновения первого признанного рецидива в течение двойного слепого периода у пациентов с AQP4-IgG
Рисунок 4. Исследование BN40900: время до возникновения первого признанного рецидива в течение двойного слепого периода у пациентов с AQP4-IgG Терапия препаратом Энспринг привела к снижению среднегодовой частоты признанных рецидивов на 74% в исследовании BN40898 и на 73% в исследовании BN40900 в сравнении с плацебо (см. таблицу 6). Относительное снижение среднегодовой частоты признанных рецидивов в подгруппе пациентов с AQP4-IgG составляло 88% и 90% в исследованиях BN40898 и BN40900 соответственно.
Таблица 6. Среднегодовая частота признанных рецидивов в течение двойного слепого периода с использованием модели отрицательной биномиальной регрессии
| BN40898 | BN40900 | Объединенные данные | ||||
| Плацебо | Энспринг | Плацебо | Энспринг | Плацебо | Энспринг | |
| ITT | N = 42 | N = 41 | N = 32 | N = 63 | N = 74 | N = 104 |
| Количество пациентов с рецидивом | 18 | 8 | 16 | 19 | 34 | 27 |
| Скорректированная среднегодовая частота рецидивов | 0.538 | 0.141 | 2.005 | 0.551 | 1.090 | 0.294 |
| Относительное снижение среднегодовой частоты признанных рецидивов (отношение рисков) | 74% (ОР: 0.261. 95% ДИ: 0.087; 0.787; p = 0.0175) | 73% (ОР: 0.275; (95% ДИ: 0.071; 1.069; p = 0.0668) | 73% (ОР: 0.270; 95% ДИ: 0.112; 0.653; p = 0.0050) | |||
| Подгруппа: Пациенты с AQP4-IgG | N = 28 | N = 27 | N = 23 | N = 41 | N = 51 | N = 68 |
| Количество пациентов с рецидивом | 12 | 3 | 13 | 9 | 25 | 12 |
| Скорректированная среднегодовая частота рецидивов | 0.520 | 0.063 | 2.853 | 0.275 | 1.339 | 0.136 |
| Относительное снижение среднегодовой частоты признанных рецидивов (отношение рисков) | 88% (ОР: 0.122. 95% ДИ: 0.027; 0.546; p = 0.0039) | 90% (ОР: 0.096. 95% ДИ: 0.020; 0.473; p = 0.0086) | 90% (ОР: 0.102; 95% ДИ: 0.034; 0.301; p = 0.0002) | |||
В сравнении с пациентами, получавшими плацебо, необходимость в неотложной терапии (например, применение глюкокортикостероидов, внутривенного иммуноглобулина и/или проведение афереза, включая плазмаферез или замещение плазмы), снизилась у пациентов, получавших препарат Энспринг, на 51% в исследовании BN40898 и на 55% в исследовании BN40900 (популяция ITT). В группе пациентов с AQP4-IgG лечение препаратом Энспринг привело к снижению необходимости применения неотложной терапии на 61% и 74% в исследованиях BN40898 и BN40900, соответственно (таблица 7).
Таблица 7. Применение неотложной терапии у пациентов с любым рецидивом, возникшим в течение двойного слепого периода
| BN40898 | BN40900 | Объединенные данные | ||||
| Плацебо | Энспринг | Плацебо | Энспринг | Плацебо | Энспринг | |
| ITT | N = 42 | N = 41 | N = 32 | N = 63 | N = 74 | N = 104 |
| Пациенты, получавшие неотложную терапию | 26 (61.90%) | 18 (43.90%) | 17 (53.13%) | 21 (33.33%) | 43 (58.11%) | 39 (37.50%) |
| Снижение риска (отношение шансов) | 51% (ОШ: 0.4915; 95% ДИ: 0.2065; 1.1698. p = 0.1084) | 55% (ОШ: 0.4509; 95% ДИ: 0.1916; 1.0612; p = 0.0682) | 54% (ОШ: 0.4649; 95% ДИ: 0.2517; 0.8589; p = 0.0145) | |||
| Подгруппа: пациенты с AQP4-IgG | N = 28 | N = 27 | N = 23 | N = 41 | N = 51 | N = 68 |
| Пациенты, получавшие неотложную терапию | 18 (64.29%) | 11 (40.74%) | 14 (60.87%) | 13 (31.71%) | 32 (62.75%) | 24 (35.29%) |
| Снижение риска (отношение шансов) | 61% (ОШ: 0.3930; 95% ДИ: 0.1343; 1.1502; p = 0.0883) | 74% (ОШ: 0.2617; 95% ДИ: 0.0862; 0.7943; p = 0.0180) | 66% (ОШ: 0.3430; 95% ДИ: 0.1614; 0.7289; p = 0.0054) | |||
Терапия препаратом Энспринг привела к снижению риска возникновения тяжелого рецидива, определяемого как увеличение EDSS на ≥2 пункта по сравнению с предыдущей оценкой по шкале EDSS, на 84% в исследовании BN40898 и на 74% в исследовании BN40900 в сравнении с плацебо (таблица 8). Относительное снижение частоты развития тяжелых рецидивов у пациентов с AQP4-IgG составляло 85% и 79% в исследованиях BN40898 и BN40900, соответственно.
Таблица 8. Время до возникновения первого тяжелого признанного рецидива в двойном слепом периоде
| BN40898 | BN40900 | Объединенные данные | ||||
| Плацебо | Энспринг | Плацебо | Энспринг | Плацебо | Энспринг | |
| ITT | N = 41 | N = 41 | N = 32 | N = 63 | N = 73 | N = 104 |
| Пациенты с событием | 6 (14.6%) | 1 (2.4%) | 6 (18.8%) | 4 (6.3%) | 12 (16.4%) | 5 (4.8%) |
| Снижение риска | 84% (ОР: 0.16; 95% ДИ: 0.02, 1.33; p = 0.0522) | 74% (ОР: 0.26; 95% ДИ: 0.07, 0.93; p = 0.0265) | 79% (ОР: 0.21; 95% ДИ: 0.07, 0.61. p = 0.0018) | |||
| Подгруппа: Пациенты с AQP4-IgG | N = 27 | N = 27 | N = 23 | N = 41 | N = 50 | N = 68 |
| Пациенты с событием | 6 (22.2%) | 1 (3.7%) | 5 (21.7%) | 3 (7.3%) | 11 (22.0%) | 4 (5.9%) |
| Снижение риска | 85% (ОР: 0.15; 95% ДИ: 0.02, 1.25; p = 0.0441) | 79% (ОР: 0.21; 95% ДИ: 0.05, 0.91; p = 0.0231) | 82% (ОР: 0.18; 95% ДИ: 0.06, 0.58; p = 0.0015) | |||
Расширенный открытый период
Анализ более долгосрочных данных, включая данные периода открытого расширенного исследования (исходя из количества рецидивов, по поводу которых пациенты получали неотложную терапию), показал, что у 57% и 71% пациентов, получавших препарат Энспринг, отсутствовали рецидивы через 120 недель терапии при применении препарата Энспринг в виде дополнительной терапии или в виде монотерапии, соответственно.
В популяции пациентов с AQP4-IgG у 58% и 73% отсутствовали рецидивы через 120 недель лечения препаратом Энспринг в виде дополнительной терапии или монотерапии, соответственно.
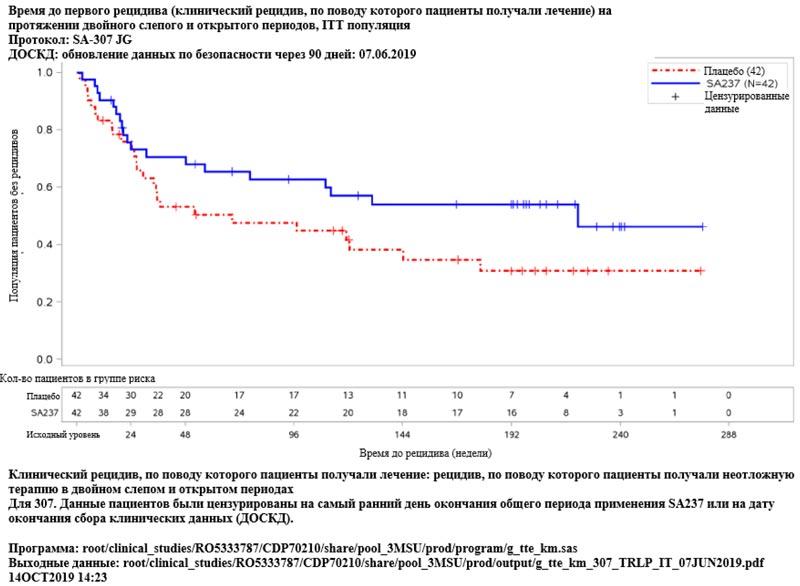 Рисунок 5. Исследование BN40898: время до первого рецидива (клинический рецидив, по поводу которого пациенты получали лечение) в двойном слепом и открытом периодах
Рисунок 5. Исследование BN40898: время до первого рецидива (клинический рецидив, по поводу которого пациенты получали лечение) в двойном слепом и открытом периодах 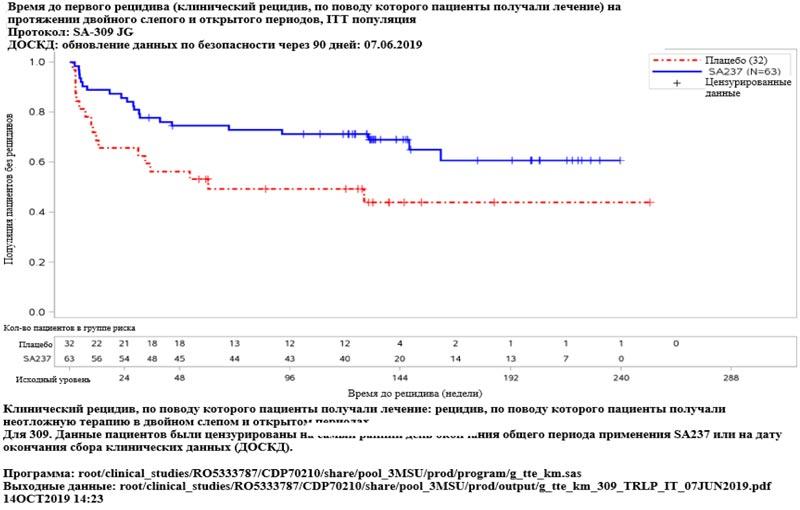 Рисунок 6. Исследование BN40900: время до первого рецидива (клинический рецидив, по поводу которого пациенты получали лечение) в двойном слепом и открытом периодах
Рисунок 6. Исследование BN40900: время до первого рецидива (клинический рецидив, по поводу которого пациенты получали лечение) в двойном слепом и открытом периодах 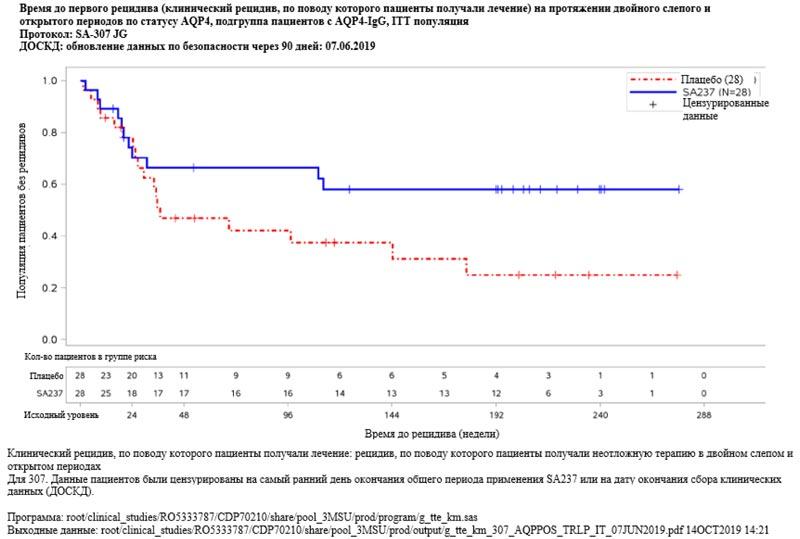 Рисунок 7. Исследование BN40898: время до первого рецидива (клинический рецидив, по поводу которого пациенты получали лечение) в течение двойного слепого и открытого периодов у пациентов с AQP4-IgG
Рисунок 7. Исследование BN40898: время до первого рецидива (клинический рецидив, по поводу которого пациенты получали лечение) в течение двойного слепого и открытого периодов у пациентов с AQP4-IgG 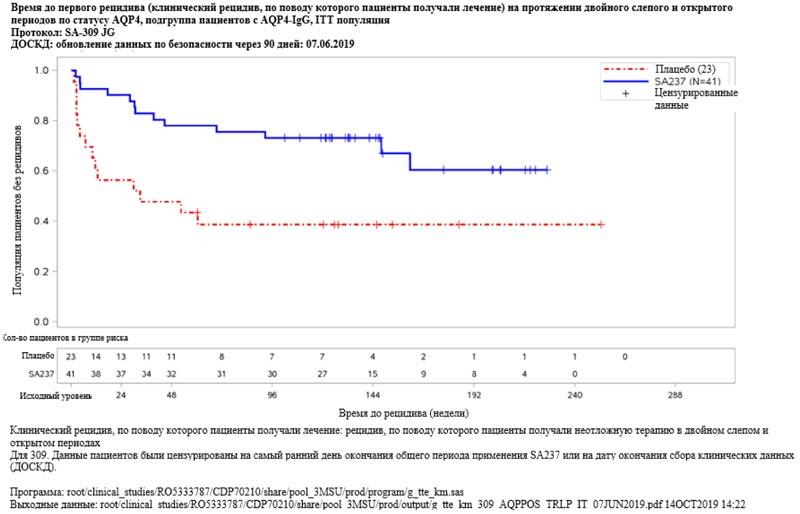 Рисунок 8. Исследование BN40900: время до первого рецидива (клинический рецидив, по поводу которого пациенты получали лечение) в течение двойного слепого и открытого периодов у пациентов с AQP4-IgG
Рисунок 8. Исследование BN40900: время до первого рецидива (клинический рецидив, по поводу которого пациенты получали лечение) в течение двойного слепого и открытого периодов у пациентов с AQP4-IgG Исходные характеристики и эффективность лечения у подростков (исследование BN40898)
Медиана возраста 7 подростков, включенных в двойной слепой период исследования BN40898, составляла 15.4 года, медиана массы тела была равна 79.6 кг. Большинство подростков возраста было женского пола (n=6). Четыре пациента являлись представителями европеоидной расы, 2 пациента были представителями негроидной расы/афроамериканцами, и 1 пациент был представителем азиатской расы. 3 из 7 подростков (42.9%) имели AQP4-IgG на момент скрининга (2 пациента в группе плацебо и 1 пациент в группе препарата Энспринг). В течение двойного слепого периода исследования у 1 из 3 подростков в группе плацебо и у 1 из 4 пациентов в группе препарата Энспринг возник признанный рецидив.
Из-за небольшого размера выборки отношение рисков для первичной конечной точки (время до возникновения первого признанного рецидива) в этой подгруппе вычислено не было.
Иммуногенность
В опорных клинических исследованиях фазы III как в комбинации с ИСТ (BN40898), так и в монотерапии (BN40900), антитела к препарату (АтП) наблюдались у 41% и 71% пациентов в двойном слепом периоде соответственно. Способность АтП нейтрализовывать связывание препарата Энспринг неизвестна.
Экспозиция препарата у пациентов с АтП была ниже, однако влияния на безопасность или явного влияния на показатели эффективности и фармакодинамики, указывающих на таргетное связывание, не было.
Терапия сатрализумабом приводила к сходному снижению риска развития признанного рецидива в опорных клинических исследованиях III фазы, независимо от различающихся показателей АтП в данных исследованиях. У пациентов с более высокой массой тела и низкой экспозицией препарата с большей вероятностью развивались АтП (независимо от фоновой ИСТ), однако эффект от лечения был сопоставим во всех группах пациентов с различной массой тела как при применении сатрализумаба в комбинации с ИСТ, так и в монотерапии. Рекомендуемая доза подходит всем пациентам, необходимость в прерывании лечения или изменения дозы у пациентов с АтП отсутствует.
5.2 Фармакокинетические свойства
Фармакокинетика препарата Энспринг была изучена у здоровых добровольцев (европеоидной расы и японцев), пациентов с оптиконевромиелитом и заболеваниями спектра оптиконевромиелита. Фармакокинетика у пациентов с оптиконевромиелитом и заболеваниями спектра оптиконевромиелита при применении препарата Энспринг в рекомендованной дозе была изучена с использованием методов анализа популяционной фармакокинетики на основании данных, полученных у 154 пациентов.
Показатель концентрация-время препарата Энспринг у пациентов с оптиконевромиелитом и заболеваниями спектра оптиконевромиелита был точно описан с помощью двухкамерной популяционной фармакокинетической модели с параллельным линейным и мишень-опосредованным (Михаэлис-Ментен) выведением и подкожным всасыванием первого порядка. Параметры клиренса и объема распределения препарата Энспринг аллометрически масштабировали в зависимости от массы тела (посредством функции мощности с зафиксированными коэффициентами мощности 0.75 и 1 для параметров клиренса и объема распределения, соответственно). Было показано, что масса тела является значимой ковариатой; при этом клиренс и центральный объем распределения у пациентов с массой тела 123 кг (97.5 процентиль распределения по массе тела) повышались на 71.3% и 105% соответственно по сравнению с пациентом с массой тела 60 кг.
Равновесное состояние показателей фармакокинетики достигалось после введения препарата в нагрузочной дозе в течение 8 недель и составило: [среднее значение (±стандартное отклонение, СО)] – 19.7 (12.2) мкг/мл для минимальной концентрации (Cmin); 31.5 (14.9) мкг/мл для максимальной концентрации (Cmax); 737 (386) мкг.мл/сутки для AUC. Применение фоновой иммунотерапии не оказывало влияние на фармакокинетику препарата (см. раздел 4.5).
Абсорбция
Константа скорости всасывания препарата Энспринг составила 0.251 1/сутки (95% ДИ: 0.216-0.285), что соответствует полупериоду всасывания (~3 дня) при применении в рекомендованной дозе (см. раздел 4.2). Биодоступность была высокой (85.4%, 95% ДИ: 79.5-95.3%).
Распределение
Препарат Энспринг подвергается двухфазному распределению. Центральный объем распределения составил 3.46 л (95% ДИ: 3.21-3.97), периферический объем распределения составил 2.07 л (95% ДИ: 1.78-2.59), интеркомпартментный клиренс – 0.336 л/сутки (95% ДИ: 0.261-0.443).
Биотрансформация
Биотрансформация препарата Энспринг специально не изучалась, поскольку моноклональные антитела выводятся, преимущественно, посредством катаболизма.
Элиминация
Общий клиренс препарата Энспринг зависит от концентрации. Установлено, что линейный клиренс составляет 0.0601 л/сутки (95% ДИ: 0.0524-0.0695; расчетное значение составляет приблизительно половину общего клиренса в равновесном состоянии при применении препарата в рекомендованной дозе у пациентов с оптиконевромиелитом и заболеваниями спектра оптиконевромиелита). Соответствующий терминальный период полувыведения (t1/2) составляет приблизительно 30 дней (диапазон 22-37 дней) согласно объединенным данным опорных клинических исследований фазы III.
Особые группы пациентов
Популяционный фармакокинетический анализ у взрослых пациентов с оптиконевромиелитом и заболеваниями спектра оптиконевромиелита показал, что возраст, пол и раса не оказывают значимого влияния на фармакокинетику сатрализумаба. Хотя масса тела оказывает влияние на фармакокинетику сатрализумаба, коррекция дозы с учетом указанных демографических факторов не рекомендуется.
Почечная недостаточность
Специального исследования по оценке влияния нарушения функции почек на фармакокинетику сатрализумаба не проводилось. Однако пациенты с нарушением функции почек легкой степени тяжести (клиренс креатинина <80 мл/мин и ≥50 мл/мин) принимали участие в клинических исследованиях BN40898 и BN40900. Как и ожидалось, на основании известного механизма клиренса сатрализумаба влияния на фармакокинетику у данных пациентов не отмечалось, таким образом, коррекции дозы не требуется.
Печеночная недостаточность
Специального исследования по изучению влияния нарушения функции печени на фармакокинетику сатрализумаба не проводилось.
Лица пожилого возраста
Отдельных исследований для изучения фармакокинетики сатрализумаба у пациентов >65 лет не проводилось, однако пациенты с оптиконевромиелитом и заболеваниями спектра оптиконевромиелита от 65 до 74 лет принимали участие в клинических исследованиях BN40898 и BN40900.
Согласно данным популяционного фармакокинетического анализа, полученных у этих пациентов, возраст не оказывает влияния на фармакокинетику сатрализумаба.
Дети
Данные, которые были получены у 8 подростков (13-17 лет) при применении препарата согласно режиму дозирования у взрослых пациентов, показали, что популяционные фармакокинетические параметры сатрализумаба у подростков не отличаются значительным образом от таковых у взрослых.
Таких образом, необходимость в коррекции дозы отсутствует.
5.3 Данные доклинической безопасности
Канцерогенность
Исследований канцерогенности с целью определения канцерогенного потенциала сатрализумаба у грызунов не проводилось. В 6-ти месячном исследовании хронической токсичности у яванских макак пролиферирующих очагов не отмечалось.
Генотоксичность
Исследований генотоксичности с целью определения мутагенного потенциала сатрализумаба не проводилось.
Не ожидается, что антитела будут оказывать влияние на ДНК.
Влияние на фертильность
При длительном введении сатрализумаба обезьянам влияния на репродуктивные органы самцов и самок не наблюдалось.
Репродуктивная токсичность
В ходе пренатального применения (до родов) сатрализумаба в дозах до 50 мг/кг 1 раз в неделю у беременных обезьян и их потомства (постнатальная экспозиция) не было выявлено какого-либо нежелательного влияния на материнский организм, развитие плода, исход беременности или выживаемость и развитие потомства, включая обучаемость.
Концентрации сатрализумаба в грудном молоке были очень низкими (<0.9% от соответствующей концентрации препарата в плазме матери).
Прочее
Токсичность при повторном введении
Доклиническое изучение применения препарата у обезьян (вид с перекрестной реактивностью к сатрализумабу) не выявило отдельных рисков для человека на основании конечных точек исследований фармакологической безопасности, острой токсичности и токсичности при повторном введении. Не наблюдалось каких-либо токсических изменений, связанных с введением сатрализумаба, при повторном подкожном введении у яванских макак в дозах до 50 мг/кг 1 раз в неделю в 4-х и 26-ти недельном исследовании токсичности.
Единственным релевантным изменением в данных исследованиях было повышение уровня IL-6 в крови, которое считается результатом фармакологического действия сатрализумаба (нейтрализующее действие в отношении IL-6R). При этом повышение уровня IL-6 в крови не было связано с какими-либо нежелательными эффектами.
Введение сатрализумаба вызывало иммунный ответ с образованием АтП у большинства животных, однако это не влияло на фармакологический ответ и не приводило к развитию каких-либо нежелательных явлений.
Местная переносимость
Применение сатрализумаба в лекарственной форме для подкожного введения не вызывало каких-либо нежелательных реакций в месте инъекции у обезьян.
Тканевая перекрестная реактивность
Тканевая перекрестная реактивность к сатрализумабу была обнаружена в тканях обезьяны и человека, экспрессирующих IL-6R. Релевантной тканевой перекрестной реактивности в других тканях обнаружено не было.
Синдром высвобождения цитокинов
На основании результатов исследований крови человека in vitro риск высвобождения провоспалительных цитокинов при применении сатрализумаба (c учетом частоты и повышения уровня цитокинов) считается низким.
6. Фармацевтические свойства
6.1 Перечень вспомогательных веществ
L-гистидин
L-аспарагиновая кислота
L-аргинин
Полоксамер 188
Вода для инъекций
6.2 Несовместимость
В связи с отсутствием исследований совместимости, данный лекарственный препарат не следует смешивать с другими лекарственными препаратами.
6.3 Срок годности (хранения)
2 года.
6.4 Особые меры предосторожности при хранении
Хранить при температуре 2-8 °С в оригинальной упаковке для защиты от света.
Не замораживать. Не использовать, если шприц-тюбик был заморожен.
Шприц-тюбики необходимо всегда сохранять сухими.
Невскрытую картонную пачку (оригинальную упаковку) можно достать и вернуть в холодильник при необходимости. Общее время хранения при комнатной температуре не выше 30 °С (вне холодильника) не должно превышать 8 дней.
6.5 Характер и содержание первичной упаковки
По 120 мг/1 мл препарата в шприц-тюбик, корпус которого изготовлен из бесцветного полимерного материала (циклоолефин, соответствует ЕФ/Ф.США/ЯФ) и силиконизирован изнутри, закрывающийся с одной стороны пластмассовым поршнем, на конце которого имеется диск из несодержащего латекс хлорбутилкаучука, ламинированного фторполимером (PTFE). С другой стороны шприц-тюбик имеет встроенную иглу для инъекций из нержавеющей стали, которая закрыта жестким защитным колпачком из несодержащего латекс хлорбутилкаучука, помещенным в наконечник из полипропилена. Шприц-тюбик со встроенной иглой помещен в защитный корпус, изготовленный из бесцветного пластика, в котором свободно вращается. Над иглой в защитном корпусе находится сжатая пружина, которая, разжимаясь после проведения инъекции, приводит в действие спусковой механизм и обеспечивает втягивание иглы внутрь защитного корпуса. На защитный корпус надет пластиковый удлиненный упор для пальцев.
1 шприц-тюбик с препаратом вместе с листком-вкладышем помещают в картонную пачку.
6.6 Особые меры предосторожности при уничтожении использованного лекарственного препарата или отходов, полученных после применения лекарственного препарата и другие манипуляции с препаратом
Шприц-тюбик с препаратом Энспринг предназначен для введения только одной дозы.
При наличии посторонних включений в растворе, его помутнении или изменении окраски не следует вводить препарат.
Необходимо проверить шприц-тюбик на предмет повреждений. При наличии трещин или повреждений не следует вводить препарат.
Не встряхивать.
Утилизация
Попадание лекарственных препаратов в окружающую среду должно быть сведено к минимуму. Не следует утилизировать препарат с помощью сточных вод или вместе с бытовыми отходами. Весь оставшийся лекарственный препарат и отходы следует уничтожить в установленном порядке.
7. Держатель регистрационного удостоверения
Швейцария
Ф. Хоффманн-Ля Рош Лтд.
F. Hoffmann-La Roche Ltd, Grenzacherstrasse 124, 4070 Basel, Switzerland
www.roche.com
7.1 Представитель держателя регистрационного удостоверения
Претензии потребителей направлять по адресу:
Республика Армения
ЗАО «Актигрупп»
0015, г. Ереван, ул. Дзорапи, д. 70/3, 4 этаж
Республика Беларусь
ИООО «Рош Продактс Лимитед»
220030, г. Минск, ул. Свердлова, д. 2, 1 этаж, помещение 20
Кыргызская Республика
Агентский офис «Ф. Хоффманн-Ля Рош Лтд.»
720055, г. Бишкек, ул. Ахунбаева 127/1, 8 этаж, каб. 808
Российская Федерация
АО «Рош-Москва»
107031, г. Москва, Трубная площадь, д. 2, помещение 1, этаж 1, комната 42
www.roche.ru
8. Номер регистрационного удостоверения
9. Дата первичной регистрации (подтверждение регистрации, перерегистрация)
Дата первой регистрации:
10. Дата пересмотра текста
Общая характеристика лекарственного препарата Энспринг доступна на информационном портале Евразийского экономического союза в информационно-коммуникационной сети «Интернет» eec.eaeunion.org.
Комментарии
ПРАКТИКА ПЕДИАТРА



