Актемра для подкожного введения - инструкция по применению
См. откуда получены инструкции МЕДИ РУ
Регистрационный номер
ЛП-003186
Торговое наименование
Актемра®
Международное непатентованное или группировочное наименование
Тоцилизумаб
Лекарственная форма
Раствор для подкожного введения
Состав
1 шприц-тюбик с раствором для подкожного введения содержит:
действующее вещество: тоцилизумаб – 162 мг;
вспомогательные вещества: полисорбат 80 – 0.18 мг, L-аргинина гидрохлорид – 19.0 мг, L-метионин – 4.03 мг, L-гистидин – 1.52 мг, L-гистидина гидрохлорида моногидрат – 1.74 мг, вода для инъекций до 0.9 мл.
Описание
Прозрачная или опалесцирующая бесцветная или желтоватого цвета жидкость.
Фармакотерапевтическая группа
Антитела моноклональные
Код АТХ
L04AC07
Фармакологические свойства
Фармакодинамика
Механизм действия
Тоцилизумаб – рекомбинантное гуманизированное моноклональное антитело к человеческому рецептору интерлейкина-6 (ИЛ-6) из подкласса иммуноглобулинов G1 (IgG1). Тоцилизумаб связывается и подавляет как растворимые, так и мембранные рецепторы ИЛ-6 (sIL-6R и mIL-6R). ИЛ-6 является многофункциональным цитокином, вырабатываемым различными типами клеток, и участвует в паракринной регуляции, системных физиологических и патологических процессах, таких как стимуляция секреции иммуноглобулинов (Ig), активация Т-клеток, стимуляция выработки белков острой фазы в печени и стимуляция гемопоэза. ИЛ-6 вовлечен в патогенез различных заболеваний, в том числе воспалительных заболеваний, остеопороза и новообразований.
Нельзя исключить вероятность отрицательного воздействия тоцилизумаба на противоопухолевую и противоинфекционную защиту организма. Роль ингибирования рецептора ИЛ-6 в развитии опухолей неизвестна.
В ходе клинических исследований тоцилизумаба у пациентов с ревматоидным артритом наблюдалось быстрое снижение уровня фибриногена.
Доклинические данные безопасности
Канцерогенность: исследования по изучению канцерогенности тоцилизумаба не проводились. Имеющиеся доклинические данные демонстрируют вклад плейотропного ИЛ-6 в прогрессирование злокачественных новообразований и устойчивость к апоптозу при различных формах рака. Эти данные не предполагают, что лечение тоцилизумабом приводит к существенному риску развития и прогрессирования рака.
Мутагенность: стандартные генотоксические тесты как в прокариотических, так и в эукариотических клетках были отрицательными.
Влияние на фертильность: имеющиеся доклинические данные не предполагают влияния аналогов тоцилизумаба на фертильность.
Тератогенность: не обнаружено прямого или опосредованного неблагоприятного влияния на беременность или внутриутробное развитие при внутривенном введении тоцилизумаба яванским макакам на ранних сроках гестационного периода.
Прочее: отмечалось незначительное увеличение случаев спонтанного выкидыша/внутриутробной гибели плода при высоком уровне системного кумулятивного воздействия (более чем в 100 раз превышающего таковое у человека) при введении дозы 50 мг/кг/сутки в сравнении с плацебо или меньшим уровнем вводимых доз. Частота выкидыша была в пределах ретроспективного контроля для яванских макак, содержащихся в неволе; отдельные случаи выкидыша/внутриутробной гибели не демонстрировали какой-либо взаимосвязи между данными явлениями и дозой или продолжительностью введения тоцилизумаба.
Несмотря на то, что ИЛ-6, по-видимому, не играет решающей роли в развитии плода или иммунологической регуляции системы мать-плод, взаимосвязь этих явлений с введением тоцилизумаба не может быть исключена.
Наблюдалась экскреция мышиного аналога тоцилизумаба в молоко лактирующих мышей.
Применение мышиного аналога тоцилизумаба не оказывало токсичного действия на ювенильных мышей. В частности, не наблюдалось нарушения роста скелета, иммунной функции и полового развития.
Доклинические данные безопасности при внутривенном применении тоцилизумаба у яванских макак не отличаются от таковых при подкожном применении тоцилизумаба.
Клиническая эффективность у пациентов с ранним ревматоидным артритом (рРА), ранее не получавших терапию метотрексатом (МТ)
При применении тоцилизумаба в монотерапии в дозе 8 мг/кг и тоцилизумаба в дозе 4 или 8 мг/кг каждые 4 недели в комбинации с МТ индекс активности заболевания по шкале DAS28 существенно снижается в группах, получавших тоцилизумаб в дозе 8 мг/кг, по сравнению с пациентами, получавшими монотерапию МТ. Число пациентов, достигших клинической ремиссии (DAS28 <2.6) на 24 неделе, значительно больше в группах, получавших тоцилизумаб, (38.7-44.8%) по сравнению с группой монотерапии МТ (15%). К 52 неделе число пациентов, достигших DAS28 <2.6 в группах терапии тоцилизумабом, увеличивается до 39.4-49% по сравнению с 19.5% в группе монотерапии МТ. Число пациентов, достигших ответа АКР 20, 50, 70 (the American College of Rheumatology, Американская Коллегия Ревматологов), также существенно выше в группах терапии тоцилизумабом (70.2-74.5%, 47.6-56.9%, 30.1-38.6% на 24 неделе и 63-67.2%, 49.3-55.9%, 36-43.1% на 52 неделе, соответственно) по сравнению с группой монотерапии МТ (65.2%, 43.2%, 25.4% на 24 неделе и 57.1%, 40.8%, 28.9% на 52 неделе, соответственно).
При монотерапии тоцилизумабом (в дозе 8 мг/кг внутривенно каждые 4 недели у пациентов с РА, c непереносимостью МТ или при нецелесообразности продолжения терапии МТ (в том числе при неадекватном ответе на терапию МТ)) наблюдалось более выраженное статистически значимое снижение активности заболевания по шкале DAS28 по сравнению с монотерапией адалимумабом (в дозе 40 мг подкожно каждые 2 недели). Количество пациентов, ответивших на терапию с показателями DAS28 <2.6 и DAS28 ≤3.2, было больше при терапии тоцилизумабом, чем при терапии адалимумабом (39.9% против 10.5% и 51.5% против 19.8%, соответственно). Ответы АКР 20, 50, 70 наблюдались у 65%, 47.2%, 32.5% пациентов, получавших тоцилизумаб, по сравнению с 49.4%, 27.8%, 17.9% пациентов, получавших адалимумаб.
Фармакокинетика
Фармакокинетика (ФК) тоцилизумаба характеризуется нелинейным выведением, представляющим комбинацию линейного клиренса и выведения по Михаэлис-Ментен. Нелинейная часть выведения тоцилизумаба приводит к более чем дозозависимому увеличению экспозиции. Фармакокинетические параметры тоцилизумаба не меняются с течением времени. Так как общий клиренс зависит от концентрации тоцилизумаба в сыворотке, период полувыведения (t½) тоцилизумаба также является зависимым от его концентрации и может быть рассчитан только при наличии значения сывороточной концентрации. Популяционный ФК анализ, проведенный во всех популяциях пациентов, не выявил связи между кажущимся клиренсом и наличием антител к тоцилизумабу.
Ревматоидный артрит (РА)
ФК тоцилизумаба сопоставима у здоровых добровольцев и пациентов с РА.
Таблица 1. Прогнозируемые средние ФК параметры (± стандартное отклонение, СО) тоцилизумаба при равновесном состоянии у пациентов с РА.
| Параметр тоцилизумаба |
162 мг 1 раз в 2 недели |
162 мг 1 раз в неделю |
| Максимальная концентрация (Cmax), мкг/мл | 13.2 ± 8.8 | 49.8 ± 21.0 |
| Минимальная концентрация (Ctrough), мкг/мл | 5.7 ± 6.8 | 43.0 ± 19.8 |
| Средняя концентрация (Cmean), мкг/мл | 10.2 ± 8.1 | 47.4 ± 20.5 |
| Кумулятивная Cmax | 2.12 | 5.27 |
| Кумулятивная Ctrough | 6.02 | 6.30 |
| Кумулятивная Cmean или площадь под кривой «концентрация-время» (AUCτ)* | 2.67 | 6.32 |
*τ - на протяжении 2 недель или 1 недели для дозы 162 мг 1 раз в 2 недели или 1 раз в неделю, соответственно.
При высоких значениях концентрации тоцилизумаба в сыворотке линейный клиренс преобладает над общим клиренсом тоцилизумаба, и конечный t½ составляет приблизительно 21.5 дней (на основе приблизительных параметров популяции пациентов). По сравнению с экспозицией тоцилизумаба при применении в дозе 162 мг подкожно (п/к) 1 раз в 2 недели экспозиция тоцилизумаба после применения в дозе 162 мг п/к 1 раз в неделю была выше в 4.6 (Cmean) и 7.5 раз (Ctrough), соответственно.
Коэффициенты накопления после многократного п/к применения тоцилизумаба во всех режимах дозирования были выше по сравнению с внутривенным (в/в) применением тоцилизумаба с максимальным значением Ctrough (6.02 и 6.30). Более высокое накопление Ctrough ожидалось благодаря участию нелинейного клиренса при более низких концентрациях тоцилизумаба.
Для Cmax более чем 90% равновесное состояние достигается после 12-ой и 5-ой п/к инъекции при введении тоцилизумаба 1 раз в неделю и 1 раз в 2 недели, соответственно; для AUCτ и Cmean 90% равновесное состояние достигается после 6-ой и 12-ой п/к инъекции при введении 162 мг тоцилизумаба 1 раз в 2 недели и 1 раз в неделю, соответственно; для Ctrough ~ 90% равновесное состояние достигается после 6-ой и 12-ой п/к инъекции тоцилизумаба для соответствующих режимов дозирования.
Доза препарата Актемра® для п/к применения не зависит от массы тела пациента с РА.
Гигантоклеточный артериит (ГА)
Таблица 2. Прогнозируемые средние ФК параметры (± стандартное отклонение, СО) тоцилизумаба при равновесном состоянии у пациентов с ГА.
| Параметр тоцилизумаба |
162 мг 1 раз в 2 недели |
162 мг 1 раз в неделю |
| Максимальная концентрация (Cmax), мкг/мл | 19.3 ± 12.8 | 73 ± 30.4 |
| Минимальная концентрация (Ctrough), мкг/мл | 11.1 ± 10.3 | 68.1 ± 29.5 |
| Средняя концентрация (Cmean), мкг/мл | 16.2 ± 11.8 | 71.3 ± 30.1 |
| Кумулятивная Cmax | 2.26 | 8.88 |
| Кумулятивная Ctrough | 5.61 | 9.59 |
| Кумулятивная Cmean или площадь под кривой «концентрация-время» (AUCτ)* | 2.81 | 10.91 |
*τ - на протяжении 2 недель или 1 недели для дозы 162 мг 1 раз в 2 недели или 1 раз в неделю, соответственно.
После применения тоцилизумаба 1 раз в неделю профиль равновесного состояния оставался практически неизменным с незначительными колебаниями средних и пиковых значений; после применения тоцилизумаба 1 раз в 2 недели наблюдались значительные колебания средних и пиковых значений. Для AUCτ приблизительно 90% равновесное состояние достигается к 14-ой неделе при введении 1 раз в 2 недели и к 17-ой неделе после введения 1 раз в неделю.
Полиартикулярный ювенильный идиопатический артрит (пЮИА)
После подкожного применения приблизительно 90% равновесное состояние достигается к 12-ой неделе при введении тоцилизумаба 1 раз в 2 недели и 1 раз в 3 недели.
Таблица 3. Прогнозируемые средние ФК параметры (± стандартное отклонение, СО) тоцилизумаба при равновесном состоянии у пациентов с пЮИА.
| Параметр тоцилизумаба |
162 мг 1 раз в 2 недели масса тела ≥30 кг |
162 мг 1 раз в 3 недели масса тела <30 кг |
| Максимальная концентрация (Cmax), мкг/мл | 29.4 ± 13.5 | 75.5 ± 24.1 |
| Минимальная концентрация (Ctrough), мкг/мл | 11.8 ± 7.08 | 18.4 ± 12.9 |
| Средняя концентрация (Cmean), мкг/мл | 21.7 ± 10.4 | 45.5 ± 19.8 |
| Кумулятивная Cmax | 1.72 | 1.32 |
| Кумулятивная Ctrough | 3.58 | 2.08 |
| Кумулятивная Cmean или площадь под кривой «концентрация-время» (AUCτ)* | 2.04 | 1.46 |
*τ - на протяжении 2 недель или 3 недель для соответствующих режимов дозирования.
Системный ювенильный идиопатический артрит (сЮИА)
После подкожного применения ~ 90% равновесное состояние достигается к 12-ой неделе при введении тоцилизумаба в дозе 162 мг 1 раз в неделю и 1 раз в 2 недели.
Таблица 4. Прогнозируемые средние ФК параметры (± стандартное отклонение, СО) тоцилизумаба при равновесном состоянии у пациентов с сЮИА.
| Параметр тоцилизумаба |
162 мг 1 раз в неделю масса тела ≥30 кг |
162 мг 1 раз в 2 недели масса тела <30 кг |
| Максимальная концентрация (Cmax), мкг/мл | 99.8 ± 46.2 | 134 ± 58.6 |
| Минимальная концентрация (Ctrough), мкг/мл | 79.2 ± 35.6 | 65.9 ± 31.3 |
| Средняя концентрация (Cmean), мкг/мл | 91.3 ± 40.4 | 101 ± 43.2 |
| Кумулятивная Cmax | 3.66 | 1.88 |
| Кумулятивная Ctrough | 4.39 | 3.21 |
| Кумулятивная Cmean или площадь под кривой «концентрация-время» (AUCτ)* | 4.28 | 2.27 |
*τ - на протяжении 1 недели или 2 недель для соответствующих режимов дозирования.
ФК параметры тоцилизумаба у пациентов в возрасте до 2-х лет были сопоставимы с ФК параметрами у пациентов старше 2-х лет с массой тела <30 кг при внутривенном применении тоцилизумаба (в дозе 12 мг/кг каждые 2 недели).
Всасывание
У пациентов с РА и ГА период полуабсорбции тоцилизумаба при п/к введении составляет около 4 дней. Биодоступность тоцилизумаба при п/к введении составляет 80%. У пациентов с ГА среднее значение времени достижения максимальной концентрации (Tmax) составляет 3 дня после применения тоцилизумаба 1 раз в неделю и 4.5 дней после применения 1 раз в 2 недели.
У пациентов с пЮИА и сЮИА период полуабсорбции тоцилизумаба при п/к введении составляет около 2-х дней, биодоступность тоцилизумаба составляет 96% и 95%, соответственно.
Распределение
Тоцилизумаб претерпевает двухфазное выведение из системного кровотока. У пациентов с РА объем распределения в центральной камере составляет 3.5 л, в периферической камере – 2.9 л, объем распределения в равновесном состоянии составляет 6.4 л.
У пациентов с ГА объем распределения в центральной камере составляет 4.09 л, в периферической камере – 3.37 л, объем распределения в равновесном состоянии составляет 7.46 л.
У детей с пЮИА объем распределения в центральной камере составляет 1.98 л, в периферической камере – 2.1 л, а объем распределения в равновесном состоянии составляет 4.08 л.
У детей с сЮИА объем распределения в центральной камере составляет 1.87 л, в периферической камере – 2.14 л, а объем распределения в равновесном состоянии составляет 4.01 л.
Выведение
Общий клиренс тоцилизумаба зависит от концентрации и представляет собой сумму линейного и нелинейного клиренса. Линейный клиренс составляет 12.5 мл/ч у пациентов с РА, 6.7 мл/ч у пациентов с ГА, 5.8 мл/ч у детей с пЮИА и 5.7 мл/ч у детей с сЮИА. Нелинейный клиренс, зависящий от концентрации, имеет наибольшее значение при низких концентрациях тоцилизумаба. При более высоких концентрациях тоцилизумаба преобладает линейный клиренс в связи с насыщением пути нелинейного клиренса. Так как общий клиренс зависит от сывороточной концентрации тоцилизумаба, период полувыведения (t½) тоцилизумаба также является зависимым от его концентрации и может быть рассчитан только при наличии значения сывороточной концентрации.
При РА зависимый от концентрации кажущийся t1/2 в равновесном состоянии составляет до 13 дней при п/к введении тоцилизумаба в дозе 162 мг 1 раз в неделю и 5 дней при п/к введении тоцилизумаба в дозе 162 мг 1 раз в 2 недели. При высоких значениях концентрации тоцилизумаба в сыворотке линейный клиренс преобладает над общим клиренсом тоцилизумаба, и конечный t½ составляет приблизительно 21.5 дней (на основе приблизительных параметров популяции пациентов).
У пациентов с ГА эффективный t½ в равновесном состоянии составляет 18.3-18.9 дней при применении тоцилизумаба в дозе 162 мг 1 раз в неделю и 4.2-7.9 дней при применении в дозе 162 мг 1 раз в 2 недели. При высоких значениях концентрации тоцилизумаба в сыворотке, когда линейный клиренс преобладает над общим клиренсом, эффективный t½ составляет приблизительно 32 дня (на основе приблизительных параметров популяции пациентов).
У детей с пЮИА эффективный t½ в равновесном состоянии составляет до 10 дней (как при применении 1 раз в 2 недели при массе тела ≥30 кг, так и 1 раз в 3 недели при массе тела <30 кг).
У детей с сЮИА эффективный t½ в равновесном состоянии составляет до 14 дней (при применении в дозе 162 мг 1 раз в неделю или 1 раз в 2 недели).
Фармакокинетика у особых групп пациентов
Пациенты с печеночной недостаточностью
Фармакокинетика тоцилизумаба у пациентов с печеночной недостаточностью не изучалась.
Пациенты с почечной недостаточностью
Специальных исследований у пациентов с почечной недостаточностью не проводилось. В ходе популяционного фармакокинетического анализа у большинства пациентов с РА и ГА наблюдалась нормальная функция почек или нарушение функции почек легкой степени тяжести (расчетный клиренс креатинина по формуле Кокрофта-Голта), которое не влияло на фармакокинетику тоцилизумаба.
Не отмечалось влияния на экспозицию тоцилизумаба у пациентов с нарушением функции почек умеренной степени тяжести (расчетный клиренс креатинина 30-59 мл/мин).
Коррекции дозы тоцилизумаба пациентам с нарушением функции почек легкой или умеренной степени тяжести не требуется.
Пол, раса, пожилой возраст
Популяционный фармакокинетический анализ у взрослых пациентов с РА и ГА показал, что возраст, пол и раса не влияют на фармакокинетику тоцилизумаба. Коррекции дозы тоцилизумаба не требуется.
Показания к применению
Ревматоидный артрит
Ревматоидный артрит со средней или высокой степенью активности у взрослых как в виде монотерапии, так и в комбинации с метотрексатом и/или с другими базисными противовоспалительными препаратами, в том числе для торможения рентгенологически доказанной деструкции суставов.
Гигантоклеточный артериит
Гигантоклеточный артериит у взрослых пациентов.
Полиартикулярный ювенильный идиопатический артрит
Активный полиартикулярный ювенильный идиопатический артрит у пациентов в возрасте 2 лет и старше как в виде монотерапии, так и в комбинации с МТ.
Системный ювенильный идиопатический артрит
Активный системный ювенильный идиопатический артрит у пациентов в возрасте 1 года и старше как в виде монотерапии, так и в комбинации с МТ.
Противопоказания
Гиперчувствительность к тоцилизумабу, любому компоненту препарата в анамнезе.
Активные инфекционные заболевания (в том числе туберкулез).
Комбинация с ингибиторами фактора некроза опухоли (ФНО)-альфа или применение в течение 1 месяца после лечения анти-ФНО антителами.
Детский возраст до 18 лет для пациентов с ревматоидным артритом и гигантоклеточным артериитом.
Детский возраст до 2 лет для пациентов с полиартикулярным ювенильным идиопатическим артритом.
Детский возраст до 1 года для пациентов с системным ювенильным идиопатическим артритом.
С осторожностью
Инфекции: у пациентов, получающих иммуносупрессанты (в том числе и препарат Актемра®), наблюдались серьезные случаи возникновения инфекционных заболеваний (иногда с летальным исходом) (см. раздел «Побочное действие»). Не следует начинать лечение препаратом Актемра® пациентам с активными инфекционными заболеваниями. При развитии серьезных инфекций терапия препаратом Актемра® должна быть прервана до разрешения инфекции. Следует соблюдать осторожность при использовании препарата Актемра® у пациентов с рецидивирующими инфекционными заболеваниями в анамнезе, а также при сопутствующих заболеваниях, предрасполагающих к развитию инфекций (например, при дивертикулите, сахарном диабете). Следует проявлять особую осторожность с целью раннего выявления серьезных инфекционных заболеваний у пациентов, получающих терапию иммуносуппрессантами (например, тоцилизумабом), поскольку признаки или симптомы острого воспаления могут быть стерты в связи с подавлением реакции острой фазы. Пациентов (включая детей раннего возраста, которые не всегда способны описать симптомы заболевания) и родителей/опекунов детей с пЮИА или сЮИА необходимо проинструктировать о немедленном обращении к врачу при любых симптомах, свидетельствующих о появлении инфекции, с целью своевременной диагностики и назначения необходимого лечения.
Осложнения дивертикулита: у пациентов, получающих терапию тоцилизумабом, отмечались случаи перфорации дивертикула. Следует соблюдать осторожность при применении препарата Актемра® у пациентов с язвенным поражением органов желудочно-кишечного тракта (ЖКТ) или дивертикулитом в анамнезе. Пациенты с признаками, возможно указывающими на осложненный дивертикулит (боль в животе), должны быть немедленно обследованы с целью раннего выявления перфорации ЖКТ.
Активные заболевания печени и печеночная недостаточность: терапия препаратом Актемра®, особенно одновременно с метотрексатом, может быть ассоциирована с повышением активности «печеночных» трансаминаз, поэтому следует проявлять осторожность у пациентов с активным заболеванием печени или печеночной недостаточностью.
Гепатотоксичность: наблюдалось легкое или умеренное повышение активности «печеночных» трансаминаз (см. раздел «Побочное действие»). Частота возникновения подобных изменений возрастала при использовании препарата Актемра® совместно с препаратами, обладающими гепатотоксическим действием (например, метотрексатом).
При применении тоцилизумаба наблюдалось серьезное лекарственное поражение печени, включая острую печеночную недостаточность, гепатит и желтуху (см. раздел «Побочное действие»). Серьезные поражения печени отмечались в интервале от 2 недель до ≥5 лет после начала терапии тоцилизумабом. Сообщалось о случаях развития печеночной недостаточности, которые привели к трансплантации печени.
Следует соблюдать осторожность при решении вопроса о начале терапии препаратом Актемра® у пациентов с показателями аланинаминотрансферазы (АЛТ) или аспартатаминотрансферазы (АСТ), превышающими верхнюю границу нормы (ВГН) более чем в 1.5 раза. Терапия препаратом Актемра® не рекомендуется при показателях АЛТ или АСТ, превышающих ВГН более чем в 5 раз. При РА, ГА, пЮИА и сЮИА следует мониторировать АЛТ/АСТ каждые 4-8 недель на протяжении первых 6 месяцев после начала терапии, а в дальнейшем – каждые 12 недель. Рекомендации по дозированию, включая отмену препарата в зависимости от активности «печеночных» трансаминаз, представлены в разделе «Способ применения и дозы».
Изменение показателей липидного обмена: наблюдалось повышение показателей липидного обмена (общего холестерина, триглицеридов и/или липопротеинов низкой плотности (ЛПНП)) (см. раздел «Побочное действие»). При терапии тоцилизумабом следует оценивать показатели липидного обмена один раз в период с 4-ой по 8-ую неделю после начала терапии препаратом Актемра®. При ведении пациентов следует руководствоваться национальными рекомендациями по лечению гиперлипидемии.
Демиелинизирующие заболевания: несмотря на то, что в настоящее время способность тоцилизумаба вызывать демиелинизирующие заболевания центральной нервной системы (ЦНС) не известна, следует проявлять особую осторожность с целью раннего выявления симптомов, возможно указывающих на развитие демиелинизирующих заболеваний ЦНС.
Применение во время беременности и в период грудного вскармливания
Беременность
Безопасность и эффективность применения препарата Актемра® при беременности изучены недостаточно. Исследования у обезьян не обнаружили дисморфогенетического потенциала препарата Актемра®. Однако при введении препарата в высоких дозах обнаружен повышенный риск спонтанного выкидыша/внутриутробной гибели. Значение данной информации для людей неизвестно (см. раздел «Фармакологические свойства», подраздел «Фармакодинамика»).
Не следует применять тоцилизумаб во время беременности, за исключением тех случаев, когда имеется очевидная клиническая необходимость.
Грудное вскармливание
Неизвестно, выводится ли препарат Актемра® с грудным молоком у человека. Несмотря на выделение эндогенных иммуноглобулинов класса G (IgG) в грудное молоко, системная абсорбция препарата при грудном вскармливании маловероятна в связи с быстрой протеолитической деградацией таких белков в системе пищеварения.
При принятии решения о продолжении/прерывании кормления грудью или продолжении/отмене терапии тоцилизумабом следует принимать во внимание пользу от грудного вскармливания для ребенка и пользу от продолжения терапии для матери.
Способ применения и дозы
Общие рекомендации
Препарат Актемра® в лекарственной форме «раствор для подкожного введения» не предназначен для внутривенного введения!
Препарат Актемра® для подкожного введения вводится с использованием одноразового шприц-тюбика.
Препарат предназначен для введения как в амбулаторно-поликлинических, так и в стационарных условиях, в том числе и для самостоятельного применения в домашних условиях. Первая инъекция должна быть произведена под наблюдением медицинского работника.
Пациент может самостоятельно производить инъекцию препарата только с разрешения врача после прохождения соответствующего обучения необходимой технике проведения инъекций и согласия на последующее медицинское наблюдение.
Рекомендуется каждый раз менять места подкожных инъекций (передняя брюшная стенка, бедро или плечо), не следует вводить препарат в места родинок, шрамов, в места повышенной чувствительности кожи, гематом, покраснения, повреждения и уплотнения кожи.
При переходе пациента с внутривенного на подкожный способ введения препарата Актемра® первую подкожную инъекцию следует произвести вместо следующей плановой внутривенной инфузии под руководством медицинского работника.
Нельзя использовать препарат при его помутнении, наличии посторонних включений и изменении окраски.
Необходимо оценить возможность самостоятельного введения препарата Актемра® самим пациентом или родителями/опекуном пациента в домашних условиях. При развитии любых симптомов серьезной аллергической реакции пациенту может потребоваться немедленная медицинская помощь. Следует проинформировать пациента или родителей/опекунов пациента о необходимости сообщать лечащему врачу о возникших симптомах аллергической реакции до проведения следующей инъекции препарата.
Ревматоидный артрит (РА)
Рекомендуемая доза препарата Актемра® для подкожного введения составляет 162 мг 1 раз в неделю. Препарат Актемра® вводится посредством подкожной инъекции и может применяться как в монотерапии, так и в комбинации с метотрексатом и/или другими базисными противовоспалительными препаратами (БПВП).
Клиническая эффективность тоцилизумаба при подкожном введении сопоставима с таковой при внутривенном введении.
Гигантоклеточный артериит (ГА)
Рекомендуемая доза препарата Актемра® для подкожного введения составляет 162 мг 1 раз в неделю. Препарат Актемра® вводится посредством подкожной инъекции в комбинации с глюкокортикостероидами с постепенным снижением дозы последних. После отмены терапии глюкокортикостероидами препарат Актемра® может применяться в монотерапии.
В случае развития обострения ГА в ходе терапии препаратом Актемра® лечащий врач должен рассмотреть возможность назначения и/или повышения дозы сопутствующей терапии глюкокортикостероидами (или повторного возобновления терапии в случае ее прерывания) согласно клиническим рекомендациям.
Рекомендации по коррекции дозы при изменении лабораторных показателей (при РА и ГА)
Повышение активности «печеночных» ферментов:
| Значение показателя | Коррекция лечения |
| Превышение ВГН* в >1-3 раза | При необходимости провести коррекцию дозы одновременно назначаемого БПВП** (РА) или иммуномодулирующих препаратов (ГА). При устойчивом повышении активности трансаминаз в данном диапазоне снизить частоту инъекций препарата Актемра® до 1 раза в 2 недели или прервать терапию препаратом Актемра® до нормализации показателей аланинаминотрансферазы (АЛТ) или аспартатаминотрансферазы (АСТ). Возобновить лечение препаратом с частотой 1 раз в неделю или 1 раз в 2 недели в соответствии с клинической необходимостью. |
| Превышение ВГН в >3-5 раз | Прервать лечение препаратом Актемра® до снижения показателя до уровня менее чем в 3 раза превышающего ВГН; далее следовать рекомендациям для превышения ВГН в >1-3 раза (см. выше). Прекратить лечение препаратом Актемра® при устойчивом повышении показателя, превышающем ВГН более чем в 3 раза (подтвержденное при повторном исследовании). |
| Превышение ВГН более чем в 5 раз | Прекратить лечение препаратом Актемра®. |
ВГН* - верхняя граница нормы
БПВП** - базисные противовоспалительные препараты
Низкое абсолютное число нейтрофилов (АЧН):
| Значение показателя (число клеток х 109/л) |
Коррекция лечения |
| АЧН >1 | Дозу не изменять. |
| АЧН 0.5-1 | Прервать лечение препаратом Актемра®. При увеличении показателя до >1 х 109/л возобновить лечение препаратом с частотой подкожных инъекций 1 раз каждые 2 недели и увеличить частоту подкожных инъекций до 1 раза в неделю в соответствии с клинической необходимостью. |
| АЧН <0.5 | Прекратить лечение препаратом Актемра®. |
Низкое число тромбоцитов:
| Значение показателя (число клеток х 103/мкл) |
Коррекция лечения |
| 50-100 | Прервать лечение препаратом Актемра®. При увеличении показателя >100 х 103/мкл возобновить лечение препаратом Актемра® с частотой подкожных инъекций 1 раз каждые 2 недели и увеличить частоту подкожных инъекций до 1 раза каждую неделю в соответствии с клинической необходимостью. |
| <50 | Прекратить лечение препаратом Актемра®. |
Полиартикулярный ювенильный идиопатический артрит (пЮИА)
Изменение дозы возможно исключительно в случае стойкого изменения массы тела пациента. Препарат Актемра® может применяться как в монотерапии, так и в комбинации с метотрексатом.
Рекомендуемая доза препарата Актемра® для подкожного введения составляет 162 мг 1 раз в 3 недели для пациентов с массой тела <30 кг и 162 мг 1 раз в 2 недели для пациентов с массой тела ≥30 кг.
Системный ювенильный идиопатический артрит (сЮИА)
Изменение дозы возможно исключительно в случае стойкого изменения массы тела пациента. Препарат Актемра® может применяться как в монотерапии, так и в комбинации с метотрексатом.
Рекомендуемая доза препарата Актемра® для подкожного введения составляет 162 мг 1 раз в 2 недели для пациентов с массой тела <30 кг и 162 мг 1 раз в неделю для пациентов с массой тела ≥30 кг. При применении препарата Актемра® в дозе 162 мг для подкожного введения пациенты в возрасте от 1 года до 2 лет должны иметь минимальную массу тела 10 кг.
Рекомендации по коррекции дозы при изменении лабораторных показателей у пациентов с пЮИА и сЮИА
Снижение дозы препарата Актемра® не изучалось у пациентов с пЮИА и сЮИА. Перерывы во введении препарата у пациентов с пЮИА или сЮИА в случае возникновения изменений лабораторных показателей рекомендуются в тех же ситуациях, которые перечислены для пациентов с РА и ГА выше (см. также разделы «Особые указания», «С осторожностью»). При необходимости следует изменить дозу одновременно принимаемого МТ и/или других сопутствующих препаратов или прекратить их прием, а также сделать перерыв во введении препарата Актемра® до разъяснения клинической ситуации. У пациентов с пЮИА или сЮИА решение о прекращении терапии препаратом Актемра® при возникновении изменений в лабораторных показателях должно быть принято в зависимости от индивидуальной клинической ситуации.
Инструкция по применению и уничтожению неиспользованного препарата и препарата с истекшим сроком годности
Попадание лекарственных препаратов вместе с отходами в окружающую среду должно быть сведено к минимуму. Не следует утилизировать препарат с помощью сточных вод или вместе с бытовыми отходами. По возможности необходимо использовать специальные системы для утилизации лекарственных препаратов.
Дозирование в особых случаях
Пожилые пациенты
Коррекции дозы у пожилых пациентов (≥65 лет) не требуется.
Пациенты с почечной недостаточностью
Коррекции дозы у пациентов с почечной недостаточностью легкой и умеренной степенями тяжести не требуется (см. раздел «Фармакологические свойства», подраздел «Фармакокинетика у особых групп пациентов»). Применение тоцилизумаба у пациентов с тяжелой почечной недостаточностью не изучалось.
Пациенты с печеночной недостаточностью
Безопасность и эффективность тоцилизумаба у пациентов с печеночной недостаточностью не изучалась (см. раздел «С осторожностью»).
Безопасность и эффективность подкожного применения тоцилизумаба у детей с пЮИА в возрасте до 2-х лет и у детей с сЮИА в возрасте до 1 года не установлены.
Дети
Инструкция по использованию шприц-тюбика
Устройство предназначено только для однократного использования.
Перед применением шприц-тюбика необходимо внимательно ознакомиться с инструкцией.
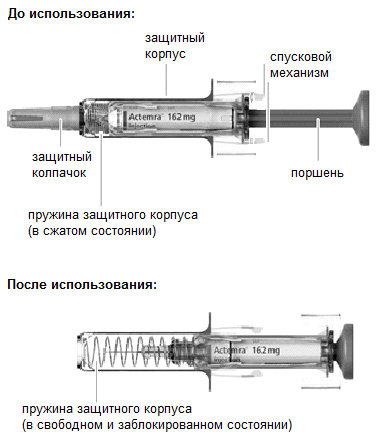
1. Осмотр шприц-тюбика
Извлеките упаковку со шприц-тюбиком из холодильника. Затем извлеките шприц-тюбик с лекарственным препаратом из картонной пачки. Осмотрите шприц-тюбик, а также лекарственный препарат, находящийся в нем.
Нельзя использовать шприц-тюбик в случае:
- помутнения раствора, наличия в препарате посторонних видимых частиц;
- изменения цвета (раствор имеет цвет, отличный от указанного в разделе «Описание»);
- повреждения любых частей шприц-тюбика;
- истечения срока годности (годен до…), указанного на картонной пачке, а также на этикетке шприц-тюбика.
Не снимайте колпачок шприц-тюбика до Этапа 5.
2. Доведение шприц-тюбика до комнатной температуры
Оставьте шприц-тюбик при комнатной температуре в течение приблизительно 25-30 минут. Не следует согревать шприц-тюбик каким-либо другим способом.
3. Обработка рук
Вымойте руки водой с мылом.
4. Выбор и подготовка места инъекции
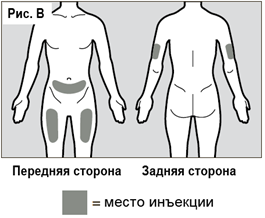
Инъекции рекомендуется производить в переднюю и среднюю поверхность середины бедра, в нижнюю часть живота, за исключением области диаметром пять сантиметров непосредственно вокруг пупка. Если инъекция производится медицинским специалистом или лицом, ухаживающим за пациентом, также возможно делать инъекции в наружную поверхность плеча (см. рис. B). Необходимо каждый раз менять место введения препарата (при проведении инъекции рекомендуется отступать не менее чем на три сантиметра от области предыдущей инъекции). Следует избегать участков, которые могут подвергаться раздражению ремнем или поясом одежды. Не следует вводить препарат в родимые пятна, ткани рубцов, гематомы, в места с уплотнением, повреждением, в участки с чувствительной кожей, покраснением и/или реакцией после предшествующих инъекций.
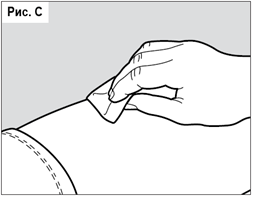
Тщательно обработайте намеченную область спиртовой салфеткой (см. рис. С). Подождите, пока обработанный участок подсохнет. Не касайтесь данной области до выполнения инъекции. Запрещается обмахивать или обдувать очищенный участок.
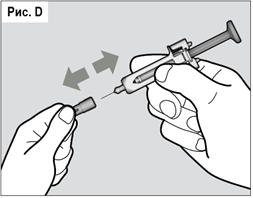
5. Подготовка шприц-тюбика
Аккуратно удерживая шприц-тюбик и не нажимая на поршень, осторожно снимите защитный колпачок с иглы (см. рис. D). После удаления колпачка следует немедленно использовать шприц-тюбик. В случае, если шприц-тюбик не был использован в течение 5 минут после снятия колпачка, он подлежит утилизации, и вместо него следует использовать новый шприц-тюбик. Не надевайте защитный колпачок после снятия.
6. Введение препарата
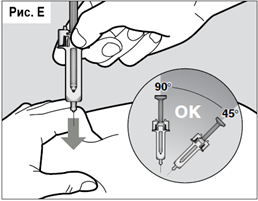
Двумя пальцами соберите кожу в складку в месте предполагаемой инъекции. Удобно удерживая шприц-тюбик другой рукой и не нажимая на поршень, введите иглу в кожную складку под углом 45-90° (угол введения зависит от толщины подкожной складки) (см. рис. Е).
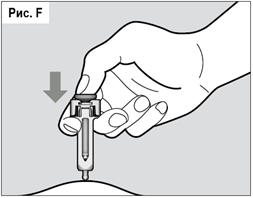
Плавно нажимая на поршень, медленно введите весь лекарственный препарат (см. рис. F). Не прекращайте давить на поршень шприц-тюбика пока полностью не извлечете иглу из кожи!
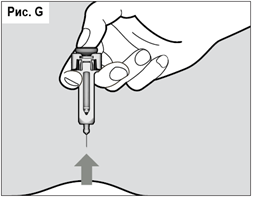
После введения всей дозы выньте иглу из кожи, не отпуская поршень шприц-тюбика (см. рис. G).
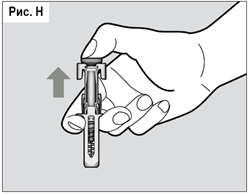
В момент отпускания поршня приводится в действие спусковой механизм и пружина, находящаяся над иглой в сжатом состоянии, освобождается (разжимается) и обеспечивает втягивание иглы внутрь защитного корпуса.
Прижмите ватным тампоном место введения лекарственного препарата. При необходимости заклейте место инъекции пластырем.
7. Утилизация шприц-тюбика
Надевать защитный колпачок на шприц-тюбик не требуется. Использованный шприц-тюбик и колпачок необходимо поместить в стойкий к прокалыванию контейнер (емкость). Данный контейнер (емкость) следует хранить в местах, недоступных для детей. Заполненный контейнер следует утилизировать в соответствии с рекомендациями медицинских специалистов. Попадание лекарственных препаратов вместе с отходами в окружающую среду должно быть сведено к минимуму. Не следует утилизировать препарат с помощью сточных вод или вместе с бытовыми отходами. По возможности необходимо использовать специальные системы для утилизации лекарственных препаратов.
Побочное действие
Для описания частоты нежелательных реакций используются следующие категории: очень часто (≥1/10), часто (≥1/100 и <1/10), нечасто (≥1/1000 и <1/100). Приведенные ниже нежелательные реакции представлены согласно классам систем органов медицинского словаря для нормативно-правовой деятельности MedDRA и перечислены в порядке клинической значимости для пациента.
Таблица 5. Обобщенные данные о нежелательных реакциях, зарегистрированных у пациентов, получавших препарат Актемра®.
| Система Орган Класс | Очень часто | Часто | Нечасто |
| Инфекции | инфекции верхних дыхательных путей | флегмона, пневмония, инфекции, вызванные Herpes simplex 1 типа и Herpes zoster | дивертикулит |
| Со стороны системы пищеварения | боли в животе, язвы ротовой полости, гастрит | стоматит, язва желудка, перфорация ЖКТ | |
| Со стороны кожи и ее придатков | сыпь, зуд, крапивница | ||
| Со стороны нервной системы | головная боль, головокружение | ||
| Изменения лабораторных показателей | повышение активности «печеночных» трансаминаз, увеличение массы тела, повышение общего билирубина | ||
| Со стороны сердечно-сосудистой системы | повышение артериального давления (АД) | ||
| Со стороны крови и лимфатической системы | лейкопения, нейтропения | ||
| Со стороны обмена веществ | гиперхолестеринемия | гипертриглицеридемия | |
| Со стороны организма в целом и реакции в месте введения | реакции в месте введения | периферические отеки, реакции гиперчувствительности | |
| Со стороны дыхательной системы | кашель, одышка | ||
| Со стороны органа зрения | конъюнктивит | ||
| Со стороны мочевыделительной системы | нефролитиаз | ||
| Со стороны эндокринной системы | гипотиреоз |
Профили безопасности тоцилизумаба у пациентов с РА, ГА, сЮИА и пЮИА сопоставимы и не подлежат дифференцировке. Ниже представлена дополнительная информация по отдельным нежелательным реакциям, наблюдавшимся в ходе клинических исследований.
Ревматоидный артрит (РА)
Профиль безопасности и иммуногенности, наблюдаемый при подкожном введении тоцилизумаба, коррелирует с известным профилем безопасности тоцилизумаба при внутривенном (в/в) введении, новых или непредвиденных нежелательных реакций не выявлено. Более высокая частота реакций в месте введения наблюдалась при подкожном применении тоцилизумаба по сравнению с подкожными инъекциями плацебо.
Профиль безопасности тоцилизумаба, исследованный у пациентов с ранним РА (рРА) средней и высокой степени активности, которые не получали ранее терапию МТ и терапию биологическими препаратами, сопоставим с известным профилем безопасности тоцилизумаба.
Реакции в месте введения: реакции в месте введения тоцилизумаба (включая эритему, зуд, боль и гематому) были легкой или средней степени тяжести, в большинстве случаев разрешались без лечения и не приводили к отмене препарата.
Иммуногенность: антитела к тоцилизумабу были выявлены у 0.8% обследованных пациентов, из них у 0.2% были выявлены иммуноглобулины класса Е (IgE). У всех пациентов выявлены нейтрализующие антитела.
Из всех 434 пациентов, получавших тоцилизумаб каждые 2 недели в дозе 162 мг, у 1.6% пациентов были выявлены антитела к тоцилизумабу, из них у 1.4% были обнаружены нейтрализующие антитела к тоцилизумабу. У 0.9% пациентов были выявлены иммуноглобулины класса Е (IgE).
Не обнаружено корреляции между наличием антител к тоцилизумабу и клиническим ответом или развитием нежелательных явлений.
Гигантоклеточный артериит (ГА)
Общий профиль безопасности тоцилизумаба у пациентов, получающих терапию препаратом Актемра®, сопоставим с известным профилем безопасности тоцилизумаба (см. Таблицу 5).
Инфекции: частота развития инфекций/серьезных инфекционных явлений была сбалансирована у пациентов, получающих тоцилизумаб 1 раз в неделю (200.2/9.7 явлений на 100 пациенто-лет), и пациентов, получающих плацебо и преднизолон в течение 26 недель (156.0/4.2 явления на 100 пациенто-лет) и 52 недель (210.2/12.5 явлений на 100 пациенто-лет), с постепенным снижением дозы преднизолона.
Полиартикулярный ювенильный идиопатический артрит (пЮИА)
Профиль безопасности тоцилизумаба у пациентов с пЮИА практически сопоставим с известным профилем безопасности тоцилизумаба, за исключением реакций в месте введения (см. Таблицу 5). У пациентов с пЮИА, получающиех п/к терапию тоцилизумабом, частота реакций в месте введения была выше, чем у взрослых пациентов с РА.
Инфекции: наиболее часто встречающееся нежелательное явление у пациентов с пЮИА. Наиболее часто встречающимися инфекциями были назофарингит и инфекции верхних дыхательных путей. Частота тяжелых инфекций, а также инфекций, приводящих к временному прекращению использования тоцилизумаба, значительно выше у пациентов с массой тела <30 кг, получавших тоцилизумаб в дозе 10 мг/кг, по сравнению с пациентами, масса тела которых была ≥30 кг, получавших тоцилизумаб в дозе 8 мг/кг. Частота развития инфекций у пациентов с пЮИА при подкожном и внутривенном введении была сопоставима.
Реакции в месте введения: отмечались у 28.8% пациентов с пЮИА. Чаще отмечались у пациентов с массой тела ≥30 кг по сравнению с пациентами с массой тела <30 кг (44% и 14.8%, соответственно). Наиболее часто встречающимися реакциями в месте введения были эритема, отек, гематома, боль и зуд. Все реакции в месте введения имели первую степень тяжести, были не серьезные и не потребовали прерывания терапии или изменения дозы тоцилизумаба.
Иммуногенность: нейтрализующие антитела к тоцилизумабу без развития серьезной или клинически значимой реакции гиперчувствительности были выявлены у 5.8% пациентов, получавших тоцилизумаб, вследствие чего 1 пациент был исключен из исследования. Связь между наличием антител и клиническим ответом на терапию или развитием нежелательных явлений не обнаружена.
Системный ювенильный идиопатический артрит (сЮИА)
В основном нежелательные реакции у пациентов с сЮИА по своему характеру не отличаются от таковых, наблюдавшихся у пациентов с РА (см. раздел «Побочное действие» выше).
Инфекции: общая частота инфекций при внутривенном введении тоцилизумаба составляла 306.6 на 100 пациенто-лет; общая частота серьезных инфекций составляла 11.3 на 100 пациенто-лет.
Частота инфекций у пациентов с сЮИА, получавших терапию тоцилизумабом п/к, была сопоставима с таковой у пациентов с сЮИА, получающих терапию тоцилизумабом в/в, а зарегистрированные серьезные инфекции не отличались от таковых у пациентов с РА, за исключением ветряной оспы и среднего отита.
Реакции в месте введения: отмечались у 41.2% пациентов с сЮИА. Наиболее часто встречающимися реакциями в месте введения были эритема, зуд, боль и отек в месте введения. Большинство реакций в месте введения имели первую степень тяжести, все реакции были не серьезные и не потребовали прерывания терапии или изменения дозы тоцилизумаба.
Иммуногенность: антитела к тоцилизумабу были выявлены у 2 из 112 обследованных пациентов. У одного из них развившаяся реакция гиперчувствительности привела к исключению из исследования. Сорок шесть (46) из 51 пациента были протестированы на наличие антител к тоцилизумабу перед началом терапии и в ходе терапии тоцилизумабом. Ни у одного пациента антитела к тоцилизумабу в ходе терапии выявлены не были.
Изменения со стороны лабораторных показателей
Четкой связи между снижением числа нейтрофилов ниже 1 х 109/л и развитием серьезных инфекционных заболеваний у пациентов с РА, ГА, пЮИА и сЮИА не отмечалось.
Ревматоидный артрит (РА)
При рутинном мониторинге лабораторных показателей отмечались следующие изменения:
- снижение числа нейтрофилов ниже 1 х 109/л отмечалось у 2.9% пациентов, получавших препарат Актемра® подкожно в дозе 162 мг 1 раз в неделю;
- снижение числа тромбоцитов ≤50 х 103/мкл не наблюдалось;
- повышение активности АЛТ или АСТ, в ≥3 раза превышающее верхнюю границу нормы, зарегистрировано у 6.5% и 1.4% пациентов, соответственно;
Транзиторное повышение активности АЛТ (более чем в 3 раза превышающее ВГН), наблюдавшееся у взрослых пациентов с рРА средней или высокой степени активности (средняя продолжительность заболевания ≤6 месяцев), которые ранее не получали терапию МТ, имело более выраженную тенденцию возврата к нормальным значениям по сравнению с популяцией пациентов с РА. - повышение показателя общего холестерина >6.2 ммоль/л (240 мг/дл) наблюдалось у 19% пациентов, а стойкое повышение показателя ЛПНП ≥4.1 ммоль/л (160 мг/дл) – у 9% пациентов.
Гигантоклеточный артериит (ГА)
При рутинном мониторинге лабораторных показателей отмечались следующие изменения:
- снижение числа нейтрофилов ниже 1 х 109/л отмечалось у 4% пациентов, получавших препарат Актемра® подкожно в дозе 162 мг 1 раз в неделю. Снижение числа нейтрофилов не отмечалось у пациентов в группе плацебо, получавших преднизолон с постепенным снижением дозы;
- однократное транзиторное снижение числа тромбоцитов <100 х 103/мкл, не сопровождающееся кровотечением, наблюдалось у 1% пациентов. Снижение числа тромбоцитов <100 х 103/мкл не отмечалось у пациентов в группе плацебо, получавших преднизолон с постепенным снижением дозы;
- повышение активности АЛТ или АСТ, в ≥3 раза (для АЛТ) и в >3 раза (для АСТ) превышающее верхнюю границу нормы, зарегистрировано у 3% и 1% пациентов, соответственно;
- повышение показателя общего холестерина >6.2 ммоль/л (240 мг/дл) наблюдалось у 29% пациентов, а стойкое повышение показателя ЛПНП ≥4.1 ммоль/л (160 мг/дл) – у 12% пациентов.
Полиартикулярный ювенильный идиопатический артрит (пЮИА)
При рутинном мониторинге лабораторных показателей отмечались следующие изменения:
- снижение числа нейтрофилов ниже 1 х 109/л отмечалось у 15.4% пациентов, получавших препарат Актемра®;
- повышение активности АЛТ или АСТ, в ≥3 раза превышающее ВГН, зарегистрировано у 9.6% и 3.8% пациентов, соответственно;
- повышение показателя ЛПНП ≥130 мг/дл отмечалось у 14.3% пациентов, получавших препарат Актемра®. Повышение показателя общего холестерина ≥200мг/дл отмечалось у 12.8% пациентов, получавших препарат Актемра®.
Системный ювенильный идиопатический артрит (сЮИА)
При рутинном мониторинге лабораторных показателей отмечались следующие изменения:
- снижение числа нейтрофилов ниже 1 х 109/л отмечалось у 23.5% пациентов, получавших препарат Актемра®;
- снижение числа тромбоцитов <100 х 109/мкл наблюдалось у 2% пациентов, получавших препарат Актемра®;
- повышение активности АЛТ или АСТ, в ≥3 раза превышающее ВГН, зарегистрировано у 9.8% и 4.0% пациентов, соответственно;
- повышение показателя ЛПНП ≥130 мг/дл и общего холестерина ≥200мг/дл отмечалось у 23.4% и 35.4% пациентов, получавших препарат Актемра®, соответственно.
Постмаркетинговое наблюдение
Приведенные ниже нежелательные реакции были выявлены в ходе постмаркетингового наблюдения на основании спонтанных сообщений, литературных источников и неинтервенционных исследовательских программ. Нежелательные реакции представлены согласно классам систем органов медицинского словаря для нормативно-правовой деятельности MedDRA. Для описания частоты нежелательных реакций используются следующие категории: часто (≥1/100 и <1/10), редко (≥1/10000 и <1/1000), очень редко (<1/10000).
Таблица 6. Нежелательные реакции, выявленные в ходе постмаркетингового применения*.
| Система Орган Класс | Часто | Редко | Очень редко |
| Со стороны кожи и ее придатков | синдром Стивенса-Джонсона1 | ||
| Со стороны крови и лимфатической системы | гипофебриногенемия | ||
| Со стороны иммунной системы | анафилаксия (фатальная)2,3 | ||
| Со стороны печени и желчевыводящих путей | лекарственное поражение печени гепатит желтуха1 |
печеночная недостаточность |
1 - реакция наблюдалась в ходе постмаркетингового наблюдения, но не отмечалась в ходе клинических исследований. Категория частоты была определена как верхняя граница 95%-го доверительного интервала, рассчитанного исходя из общего количества пациентов, получавших тоцилизумаб в клинических исследованиях.
2 - см. раздел «Особые указания».
3 - см. раздел «Противопоказания».
* - частота встречаемости рассчитывается на основании данных, полученных из соответствующих завершенных клинических исследований по всем показаниям.
Передозировка
Данные о передозировке препарата Актемра® ограничены. В одном случае непреднамеренной передозировки препарата в дозе 40 мг/кг внутривенно у пациента с множественной миеломой нежелательных реакций не отмечено. Не отмечалось также серьезных нежелательных реакций у здоровых добровольцев, которые получали однократно препарат Актемра® в дозе до 28 мг/кг внутривенно, хотя наблюдалась нейтропения, требующая снижения дозы.
Взаимодействие с другими лекарственными средствами
Популяционный фармакокинетический анализ клинических исследований не выявил какого-либо воздействия метотрексата, нестероидных противовоспалительных препаратов или глюкокортикостероидов на клиренс тоцилизумаба у пациентов с РА. У пациентов с ГА кумулятивные дозы глюкокортикостероидов не оказывали влияния на экспозицию тоцилизумаба.
Фармакокинетические параметры тоцилизумаба остаются неизменными при одновременном применении других противоревматических препаратов (таких как противомалярийные препараты (хлорохин и его производные), иммуносупрессанты (азатиоприн, лефлуномид), фолиевая кислота и ее производные, ингибиторы циклооксигеназы-2 (целекоксиб) и анальгетики (парацетамол, трамадол, кодеин и их производные)).
Одновременное однократное введение тоцилизумаба в дозе 10 мг/кг и метотрексата в дозе 10-25 мг один раз в неделю не оказывало клинически значимого влияния на экспозицию метотрексата.
Исследования по изучению комбинированного применения тоцилизумаба с другими биологическими БПВП не проводились.
Поскольку экспрессия печеночных изоферментов CYP450 подавляется под действием цитокинов (например, ИЛ-6, который стимулирует хроническое воспаление), при проведении терапии средствами, ингибирующими действие цитокинов (например, тоцилизумаб), экспрессия изоферментов CYP450 может быть нарушена. В исследованиях in vitro, проведенных на культуре гепатоцитов человека, было показано, что ИЛ-6 вызывает снижение экспрессии изоферментов CYP1A2, CYP2C9, CYP2C19 и CYP3A4. Применение тоцилизумаба нормализует экспрессию этих изоферментов. Влияние препарата Актемра® на изоферменты CYP (кроме СYP2C19 и CYP2D6) имеет клиническое значение для препаратов, являющихся субстратами CYP450, с узким терапевтическим индексом и/или для которых дозы подбираются индивидуально. У пациентов с ревматоидным артритом концентрация симвастатина (субстрат СYP3A4) через 1 неделю после однократного введения тоцилизумаба снижалась на 57%, т.е. была немного повышенной или аналогичной таковой у здоровых добровольцев. В начале или при завершении курса терапии препаратом Актемра® следует тщательно наблюдать за пациентами, получающими лекарственные средства в индивидуально подобранных дозах, и которые метаболизируются посредством изоферментов CYP450 3A4, 1A2 или 2C9 (например, аторвастатин, блокаторы «медленных» кальциевых каналов, теофиллин, варфарин, фенитоин, циклоспорин или бензодиазепины). Для обеспечения терапевтического действия этих препаратов возможно потребуется увеличение их дозы. Учитывая длительный t½ препарата Актемра®, его действие на активность CYP450 ферментов может сохраняться в течение нескольких недель после прекращения терапии.
Особые указания
В медицинской документации пациента следует указывать торговое наименование препарата и номер серии. Замена препарата Актемра® для подкожного введения на какой-либо другой биологический лекарственный препарат требует согласования с лечащим врачом.
Реакции гиперчувствительности: при постмаркетинговом применении препарата Актемра® для внутривенного введения наблюдались серьезные реакции гиперчувствительности, включая анафилаксию (см. раздел «Побочное действие»). Данные серьезные явления могут являться тяжелыми и потенциально фатальными у пациентов, ранее имевших реакции гиперчувствительности при применении препарата Актемра® в комплексе с премедикацией глюкокортикостероидами и антигистаминными препаратами.
При введении препарата Актемра® должен быть предусмотрен комплекс необходимых мероприятий для лечения возможной анафилактической реакции. При возникновении анафилактической реакции или другой серьезной реакции гиперчувствительности введение препарата Актемра® следует немедленно прекратить, начать соответствующее лечение и не возобновлять терапию препаратом в дальнейшем.
Туберкулез: до назначения препарата Актемра®, как и при назначении других биологических препаратов, следует провести предварительное обследование пациентов на наличие латентного туберкулеза. При выявлении латентного туберкулеза следует провести стандартный курс антимикобактериальной терапии перед началом лечения препаратом Актемра®.
Иммунизация: не следует проводить иммунизацию живыми и живыми ослабленными вакцинами одновременно с терапией препаратом Актемра®, поскольку безопасность подобного сочетания не установлена. Отсутствуют данные о вторичной передаче инфекции от пациентов, получающих живые вакцины, к пациентам, получающим тоцилизумаб. У пациентов с ревматоидным артритом, получающих терапию тоцилизумабом/ метотрексатом, ответ на 23-валентную пневмококковую полисахаридную вакцину и столбнячный анатоксин был сопоставим с таковым у пациентов, получающих монотерапию метотрексатом. Рекомендуется, чтобы до начала лечения препаратом Актемра® все пациенты (особенно дети и пациенты пожилого возраста) прошли вакцинацию в соответствии с национальным календарем прививок. Следует соблюдать интервал (в соответствии с действующими рекомендациями по иммунизации) у пациентов, получающих терапию иммуносупрессивными препаратами, между иммунизацией живыми вакцинами и началом терапии препаратом Актемра®.
Реактивация вирусных инфекций: у пациентов с ревматоидным артритом, получавших терапию биологическими препаратами, наблюдались случаи реактивации вирусной инфекции (например, вирусного гепатита В). Пациенты, имевшие положительный результат при скрининговом обследовании на гепатит, не включались в клинические исследования препарата Актемра®.
Синдром активации макрофагов у пациентов с сЮИА
Синдром активации макрофагов является серьезным жизнеугрожающим состоянием, которое может развиться у пациентов с сЮИА. Эффективность и безопасность препарата Актемра® в период возникновения синдрома активации макрофагов не изучались.
Изменения лабораторных показателей
Нейтропения: терапия препаратом Актемра® ассоциировалась с более высокой частотой развития нейтропении. Нейтропения, связанная с лечением, не ассоциировалась с развитием серьезных инфекций (см. раздел «Побочное действие»). Следует проявлять осторожность при назначении препарата Актемра® пациентам с нейтропенией, т.е. при абсолютном числе нейтрофилов (АЧН) <2.0 х 109/л. При АЧН <0.5 x 109/л лечение препаратом Актемра® не рекомендуется. При РА и ГА следует мониторировать число нейтрофилов. Первое определение количества нейтрофилов при РА и ГА рекомендуется выполнить в период с 4-ой по 8-ую неделю после начала терапии, а в дальнейшем в соответствии с клинической практикой. Рекомендации по дозированию препарата в зависимости от АЧН представлены в разделе «Способ применения и дозы».
При пЮИА и сЮИА число нейтрофилов следует контролировать в день проведения 2-го введения, а в дальнейшем в соответствии с клинической практикой (см. раздел «Способ применения и дозы»).
Тромбоцитопения: терапия препаратом Актемра® ассоциировалась со снижением числа тромбоцитов. Снижение числа тромбоцитов, связанное с лечением, не ассоциировалось с серьезными случаями кровотечений (см. раздел «Побочное действие»). Следует соблюдать осторожность при решении вопроса о начале терапии препаратом Актемра® при числе тромбоцитов ниже 100 х 103/мкл. Лечение не рекомендуется при числе тромбоцитов <50 х 103/мкл. При РА и ГА следует мониторировать число тромбоцитов первый раз в период с 4-ой по 8-ую неделю после начала терапии, а в дальнейшем в соответствии с клинической практикой. Рекомендации по дозированию препарата в зависимости от числа тромбоцитов представлены в разделе «Способ применения и дозы».
При пЮИА и сЮИА число тромбоцитов следует мониторировать в день проведения 2-го введения, а в дальнейшем в соответствии с клинической практикой (см. раздел «Способ применения и дозы»).
Особенности действия лекарственного препарата при первом приеме или при его отмене
Исследования по изучению возможности препарата Актемра® вызывать зависимость не проводились. Исходя из имеющихся данных, препарат Актемра® не обладает таким действием.
Влияние на способность управлять транспортными средствами и механизмами
Исследования по изучению влияния препарата на способность управлять транспортными средствами и механизмами не проводились. Однако, учитывая тот факт, что при терапии препаратом Актемра® часто наблюдалось головокружение, пациентам, испытывающим данную нежелательную реакцию, следует рекомендовать не управлять транспортными средствами и механизмами до тех пор, пока головокружение не прекратится.
Форма выпуска
Раствор для подкожного введения 162 мг/0.9 мл
По 162 мг/0.9 мл препарата в шприц-тюбик, корпус которого изготовлен из нейтрального стекла типа 1 (ЕФ), закрывающийся с одной стороны поршнем, изготовленным из пластмассы, на конце которого имеется диск из бутилкаучука, ламинированного фторполимером. С другой стороны шприц-тюбик имеет встроенную иглу для инъекций, которая закрыта колпачком из не содержащего латекс полиизопренового каучука, помещенного в полимерный наконечник. Шприц-тюбик со встроенной иглой помещен в защитный корпус, изготовленный из бесцветного пластика, в котором свободно вращается. Над иглой в защитном корпусе находится сжатая пружина, которая, разжимаясь после проведения инъекции, приводит в действие спусковой механизм и обеспечивает втягивание иглы внутрь защитного корпуса.
4 шприц-тюбика вместе с инструкцией по применению помещают в картонную пачку.
Условия хранения
Хранить при температуре 2-8 °C в защищенном от света месте.
Не замораживать.
Хранить в недоступном для детей месте.
Срок годности
2 года.
Не использовать по истечении срока годности, указанного на упаковке.
Условия отпуска
По рецепту.
Владелец Регистрационного удостоверения
Ф. Хоффманн-Ля Рош Лтд., Швейцария
F. Hoffmann-La Roche Ltd, Grenzacherstrasse 124, 4070 Basel, Switzerland
Производитель
Веттер Фарма-Фертигунг ГмбХ и Ко. КГ, Германия
Vetter Pharma-Fertigung GmbH & Co. KG, Schuetzenstrasse 87 und 99-101, 88212 Ravensburg, Germany
Организация, уполномоченная на принятие претензий от потребителей
Претензии потребителей направлять в компанию АО «Рош-Москва» по адресу:
107045, Россия, г. Москва, Трубная площадь, д. 2, помещение 1, этаж 1, комната 42
тел. (495) 229 29 99, факс (495) 229 79 99
или через форму обратной связи на сайте:
Комментарии
ПРАКТИКА ПЕДИАТРА



