Афстила - инструкция по применению
См. откуда получены инструкции МЕДИ РУ
Регистрационный номер:
ЛП-006975
Торговое наименование:
Афстила®
Международное непатентованное наименование:
лоноктоког альфа (фактор свертывания крови VIII человеческий одноцепочечный)
Лекарственная форма:
лиофилизат для приготовления раствора для внутривенного введения (в комплекте с растворителем вода для инъекций).
Состав
на 1 флакон с лиофилизатом:
| Лиофилизат | 250 ME | 500 ME | 1000 ME | 1500 ME | 2000 ME | 2500 ME | 3000 ME |
| Действующее вещество | |||||||
| Лоноктоког альфа | 250 ME | 500 ME | 1000 ME | 1500 ME | 2000 ME | 2500 ME | 3000 ME |
| Вспомогательные вещества | |||||||
| L-гистидин | 7,8 мг | 7,8 мг | 7,8 мг | 15,5 мг | 15,5 мг | 15,5 мг | 15,5 мг |
| Сахароза | 15,0 мг | 15,0 мг | 15,0 мг | 30,0 мг | 30,0 мг | 30,0 мг | 30,0 мг |
| Полисорбат 80 | 0,5 мг | 0,5 мг | 0,5 мг | 1,0 мг | 1,0 мг | 1,0 мг | 1,0 мг |
| Кальция хлорид | 0,9 мг | 0,9 мг | 0,9 мг | 1,9 мг | 1,9 мг | 1,9 мг | 1,9 мг |
| Натрия хлорид | 41,0 мг | 41,0 мг | 41,0 мг | 82,0 мг | 82,0 мг | 82,0 мг | 82,0 мг |
| Кислота хлористоводородная | q.s. для доведения pH | ||||||
Растворитель
Вода для инъекций 2,5 мл 2,5 мл 2,5 мл 5,0 мл 5,0 мл 5,0 мл 5,0 мл
После восстановления 2,5 мл воды для инъекций (дозировок 250/500/1000 ME) раствор содержит 100/200/400 МЕ/мл фактора свертывания крови VIII человеческого одноцепочечного соответственно.
После восстановления 5,0 мл воды для инъекций (дозировок 1500/2000/2500/3000 ME) раствор содержит 300/400/500/600 МЕ/мл фактора свертывания крови VIII человеческого одноцепочечного соответственно.
Фармакотерапевтическая группа:
гемостатическое средство.
Код ATX:
B02BD02.
Описание:
Лиофилизат: порошок или хрупкая масса от белого до слегка желтого цвета.
Восстановленный раствор: почти бесцветная жидкость от прозрачной до слегка опалесцирующей.
Растворитель: бесцветная прозрачная жидкость.
Фармакологические свойства:
Действующее вещество препарата Афстила® (лоноктоког альфа) представляет собой фактор свертывания крови VIII человеческий одноцепочечный, полученный методом генной инженерии в клетках СНО, необходимый для эффективного гемостаза при гемофилии А. Лоноктоког альфа представляет собой полипептидную цепь с усеченным В-доменом, обеспечивающим ковалентную связь между тяжелой и легкой цепями фактора свертывания крови VIII. Он обладает более высокой аффинностью к фактору фон Виллебранда, чем полноразмерный фактор свертывания крови VIII человеческий рекомбинантный. Фактор фон Виллебранда стабилизирует фактор свертывания крови VIII и защищает его от разрушения. Функциональные характеристики фактора свертывания крови VIII человеческого одноцепочечного сходны с таковыми эндогенного фактора свертывания крови VIII, активность которого значительно снижена у пациентов с гемофилией А, что требует проведения заместительной терапии.
Фармакодинамика
Гемофилия А это сцепленное с полом наследственное нарушение свертывания крови в связи с пониженным уровнем фактора свертывания крови VIII, приводящее к массивным кровоизлияниям в суставы, мышцы и внутренние органы, как спонтанно, так и в результате травм и оперативных вмешательств. При проведении заместительной терапии уровень фактора свертывания крови VIII в плазме повышается, в результате чего происходит временная коррекция дефицита фактора в плазме крови и снижается тенденция к повышенной кровоточивости.
Комплекс фактор свертывания крови VIII/фактор фон Виллебранда состоит из двух молекул (фактор свертывания крови VIII и фактор фон Виллебранда) с различными физиологическими функциями. При внутривенном введении пациентам с гемофилией фактор свертывания крови VIII связывается с эндогенным фактором Виллебранда в системе кровообращения пациента. Активированный фактор свертывания крови VIII действует как ко-фактор для активированного фактора свертывания крови IX, ускоряя преобразование фактора свертывания крови X в активированный фактор свертывания крови X. Активированный фактор свертывания крови X способствует переходу протромбина в тромбин. Тромбин в свою очередь способствует переходу фибриногена в фибрин, что приводит к образованию тромба.
Взрослые и подростки в возрасте от 12 до 65 лет
Было проведено клиническое исследование эффективности и безопасности при применении препарата Афстила® в режиме профилактики, с целью предотвращения кровотечений, эффективности гемостаза при введении с целью контроля кровотечений и в периоперационном периоде среди 175 пациентов с тяжелой гемофилией А, ранее получавших лечение. Ни у одного пациента не наблюдалось образования ингибиторов или развития анафилактических реакций.
Дети в возрасте младше 12 лет
В клиническом исследовании с участием 84 пациентов в возрасте младше 12 лет, ранее получавших лечение, ни у одного пациента не наблюдалось образования ингибиторов или развития анафилактических реакций.
Фармакокинетика
Взрослые пациенты
Фармакокинетику препарата Афстила® оценивали у 81 ранее получавших лечение пациента в возрасте от 18 до 60 лет с диагнозом тяжелая гемофилия А (исходный уровень фактора свертывания крови VIII <1%) после внутривенного введения дозы 50 МЕ/кг препарата. Оценка фармакокинетических показателей была основана на активности фактора свертывания крови VIII в плазме крови, измеряемой с помощью анализа с использованием хромогенного субстрата (при использовании одноступенчатого анализа коагуляционной активности следует умножить результат на коэффициент пересчета 2, чтобы определить активность фактора свертывания крови VIII пациента). Фармакокинетический профиль, полученный через 3-6 месяцев многократного введения препарата Афстила®, был сопоставим с фармакокинетическим профилем, полученным после введения первой дозы.
Таблица 1. Фармакокинетические параметры препарата Афстила® у пациентов с тяжелой гемофилией А после однократной инъекции дозы 50 МЕ/кг измеренные с помощью анализа с использованием хромогенного субстрата
| Фармакокинетические показатели | Фактор свертывания крови VIII человеческий одноцепочечный Доза 50 МЕ/кг (N=81) Среднее (КВ%), Медиана (Мин, Макс) |
| СН (МЕ/дл)/(МЕ/кг) | 2,00 (20,8) 1,99 (0,868; 2,90) |
| Сmax (МЕ/ДЛ) | 106 (18,1) 106 (62,4; 151) |
| AUC0-inf (МЕ*ч/дл) | 1960 (33,1) 1910 (932,4090) |
| t1/2 (ч) | 14,2 (26,0) 13,7 (7,54; 23,9) |
| Среднее время циркуляции в плазме (ч) | 20,4 (25,8) 20,2 (10,8; 35,1) |
| Клиренс (мл/ч/кг) | 2,90 (34,4) 2,67 (1,26; 5,79) |
| Vss (мл/кг) | 55,2 (20,8) 53,2 (32,4; 99,6) |
СН = Скорректированное нарастание восстановления через 30 мин после введения; Сmax = максимальная концентрация, AUC0-inf = площадь под кривой зависимости активности фактора свертывания крови VIII от времени, экстраполированная до бесконечности; t1/2 = период полувыведения; клиренс = клиренс, скорректированный с учётом массы тела для N=80; Vss = объем распределения в равновесном состоянии. СН и Сmax были скорректированы по базовой линии, в то время как остальные параметры не были скорректированы по базовой линии для N=81.
Дети
Фармакокинетику препарата Афстила® оценивали у 10 ранее получавших лечение подростков (в возрасте от 12 до 18 лет) и у 39 ранее получавших лечение детей (в возрасте от 0 до 12 лет) после внутривенного введения однократной дозы 50 МЕ/кг. Все пациенты имели диагноз тяжелая гемофилия А с исходным фактором свертывания крови VIII <1%.
Фармакокинетические показатели были основаны на активности фактора свертывания крови VIII в плазме крови, измеренной с помощью анализа с использованием хромогенного субстрата (при использовании одноступенчатого анализа коагуляционной активности следует умножить результат на коэффициент пересчета 2, чтобы определить активность фактора свертывания крови VIII пациента).
Таблица 2. Сравнение фармакокинетических параметров препарата Афстила® у пациентов с тяжелой гемофилией А после однократной инъекции 50 МЕ/кг в зависимости от возраста, измеренные с помощью анализа с использованием хромогенного субстрата
| Фармакокинетические показатели | от 0 до <6 лет (N=20) среднее (КВ%), медиана (мин, макс) | от 6 до <12 лет (N=19) среднее (КВ%), медиана (мин, макс) | от 12 до <18 лет (N=10) среднее (КВ%), медиана (мин, макс) |
| СН (МЕ/дл)/(МЕ/кг) | 1,60 (21,1) 1,55 (1,18; 2,76) | 1,66(19,7) 1,69 (0,92; 2,35) | 1,69 (24,8) 1,76 (0,88; 2,44) |
| Сmax (МЕ/ДЛ) | 80,2 (20,6) 78,6 (59,3; 138) | 83,5(19,5) 84,5 (46,4; 117) | 89,7 (24,8) 92,4 (45,5; 131) |
| AUC0-inf (МЕ*ч/дл) | 1080 (31,0) 985 (561,2010) | 1170 (26,3) 1120 (641,1810) | 1540 (36,5) 1520 (683, 380) |
| t1/2 (ч) | 10,4 (28,7) 10,1 (5,19; 17,8) | 10,2(19,4) 10,0 (6,92; 14,8) | 14,3 (33,3) 13,5 (6,32; 3,8) |
| Среднее время циркуляции в плазме (ч) | 12,4 (25,0) 13,0 (6,05; 17,9) | 12,3 (16,8) 12,8 (8,22; 16,0) | 20,0 (32,2) 18,6 (9,17; 31,7) |
| Клиренс (мл/ч/кг) | 5,07 (29,6) 5,08 (2,52; 8,92) | 4,63 (29,5) 4,48 (2,79; 7,71) | 3,80 (46,9) 3,31 (2,10; 7,32) |
| Vss (мл/кг) | 71,0 (11,8) 70,7 (57,3; 88,3) | 67,1 (22,3) 64,9 (44,3; 111) | 68,5 (29,9) 62,0 (45,9; 121) |
СН = Скорректированное нарастание восстановления через 30 мин после введения для пациентов в возрасте от 12 до <18 лет и через 60 мин после введения для пациентов в возрасте от 1 до <12 лет; Сmax = максимальная концентрация, AUC0-inf = площадь под кривой зависимости активности фактора свертывания крови VIII от времени, экстраполированная до бесконечности; t1/2 = период полувыведения; клиренс = клиренс, скорректированный с учётом массы тела; Vss = объем распределения в равновесном состоянии. СН и Сmax были скорректированы по базовой линии, в то время как остальные параметры не были скорректированы по базовой линии.
По сравнению с подростками и взрослыми у детей в возрасте до 12 лет может быть более высокий клиренс и более короткий период полувыведения, что согласуется с данными по другим препаратам факторов свертывания. Следует принимать во внимание эти различия при дозировании препарата.
Показания к применению:
Лечение и профилактика кровотечений у пациентов с гемофилией А (врожденной недостаточностью фактора свертывания крови VIII), ранее получавших заместительную терапию.
Препарат Афстила® показан к применению во всех возрастных группах.
Противопоказания:
Гиперчувствительность к лоноктокогу альфа или к любому из компонентов, входящих в состав препарата.
Гиперчувствительность к белкам животного происхождения в анамнезе.
С осторожностью:
Клинических исследований эффективности и безопасности препарата Афстила® у пациентов в возрасте старше 65 лет не проводилось, поэтому рекомендуется соблюдать осторожность при применении препарата Афстила® у таких пациентов.
Применение при беременности и в период грудного вскармливания:
Доклинических исследований репродуктивной токсичности фактора свёртывания крови VIII не проводилось. Учитывая, что гемофилия типа А очень редко наблюдается у женщин, отсутствуют данные об опыте применения препаратов фактора свертывания крови VIII во время беременности и в период грудного вскармливания. В связи с этим препарат Афстила® следует применять в период беременности или грудного вскармливания только при наличии строгих показаний.
Способ применения и дозы:
Только для внутривенного введения! Терапия препаратом Афстила® должна проводиться врачами, имеющими опыт лечения пациентов с гемофилией. Препарат вводится внутривенно в течение нескольких минут после восстановления лиофилизата прилагаемым растворителем (см. Инструкцию по приготовлению раствора для внутривенного введения). Восстановленный препарат следует вводить медленно со скоростью, приемлемой для пациента, не превышающей 10 мл/мин. Расчет требуемой дозы фактора свертывания крови VIII основан на эмпирических данных о том, что 1 международная единица (ME) фактора свертывания крови VIII на кг массы тела повышает активность этого фактора в плазме крови на 2 МЕ/дл. Необходимую дозу рассчитывают по следующей формуле:
Доза (ME) = масса тела (кг) х желаемое повышение уровня фактора свертывания крови VIII (МЕ/дл или% от нормы) х 0,5 (МЕ/кг на МЕ/дл).
При выборе дозы и частоты введения препарата следует всегда ориентироваться на клиническую эффективность в каждом индивидуальном случае.
Контроль лечения
В ходе лечения рекомендуется определять уровень фактора свертывания крови VIII для коррекции вводимой дозы и частоты повторных инъекций. Ответ на введение фактора свертывания крови VIII у отдельных пациентов может варьироваться, демонстрируя различные периоды полувыведения и восстановления. Доза, рассчитанная по массе тела, может потребовать корректировки у пациентов с недостаточной или избыточной массой тела. В случае обширных хирургических вмешательств тщательный контроль заместительной терапии посредством анализа свертываемости крови (активность фактора свертывания крови VIII в плазме крови) является обязательным.
При использовании одноступенчатого определения in vitro активированного частичного тромбопластинового времени (АЧТВ) для оценки активности фактора свертывания крови VIII в образцах крови пациентов результаты активности фактора свертывания крови VIII в плазме могут в значительной степени зависеть от используемых в каждом конкретном случае определения АЧТВ реагентов и референтных стандартов. Кроме того, может иметь место значительное расхождение между результатами одноступенчатого теста коагуляционной активности с использованием АЧТВ-реагента и хромогенного анализа. Это имеет особое значение при смене лаборатории и/или реагентов, используемых при проведении тестов.
У пациентов, получающих препарат Афстила®, для контроля вводимой дозы и частоты повторных инъекций необходимо определять активность фактора свертывания крови VIII в плазме крови с использованием хромогенного анализа либо одноступенчатого теста коагуляционной активности. Заявляемая активность препарата Афстила® основана на результатах хромогенного теста. При проведении мониторинга лечения рекомендуется использовать хромогенный тест. Результаты, полученные с помощью этого теста, как правило выше, чем при использовании одноэтапного анализа коагуляционной активности (примерно на 45%). С целью коррекции данных несоответствий результаты одноэтапного коагуляционного теста следует умножить на 2.
Режим дозирования
Дозы и продолжительность заместительной терапии препаратом Афстила® зависят от степени тяжести дефицита фактора свертывания крови VIII, локализации и выраженности кровотечения, а также от клинического состояния пациента. Количество вводимых единиц фактора свертывания крови VIII измеряется в международных единицах (ME), что соответствует действующему стандарту Всемирной Организации Здравоохранения (ВОЗ) для препаратов, содержащих фактор свертывания крови VIII. Активность фактора свертывания крови VIII в плазме выражается в процентах (относительно нормальной плазмы человека) или в ME (относительно международного стандарта содержания фактора свертывания крови VIII в плазме).
Одна ME активности фактора свертывания крови VIII эквивалентна количеству фактора свертывания крови VIII в 1 мл нормальной плазмы человека.
Оценку активности проводят с помощью анализа с использованием хромогенного субстрата.
Уровень фактора свертывания крови VIII в плазме крови можно контролировать с помощью анализа с использованием хромогенного субстрата или одноступенчатого анализа коагуляционной активности.
Лечение по требованию
Расчет необходимой дозы фактора свертывания крови VIII основывается на эмпирически определенной закономерности, согласно которой 1 ME фактора свертывания крови VIII на килограмм массы тела повышает активность этого фактора в крови на 2,0 МЕ/дл. Необходимую дозу рассчитывают с использованием следующей формулы:
Доза (ME) = масса тела (кг) × требуемое повышение фактора свертывания крови VIII (МЕ/дл или% от нормы) × 0,5 (МЕ/кг на МЕ/дл)
Вводимая доза и частота введения должны рассчитываться с учетом клинической эффективности в каждом индивидуальном случае.
В случаях кровотечений, приведенных в Таблице 3, активность фактора свертывания крови VIII за соответствующий период времени не должна быть ниже указанного в таблице уровня активности в плазме (в% от нормального уровня или МЕ/дл). Таблица 3 может быть использована для расчета доз препарата при кровотечениях и в хирургической практике.
Таблица 3. Требуемый уровень активности фактора VIII для лечения по требованию при кровотечениях и при хирургических вмешательствах
| Тяжесть кровотечения / тип хирургического вмешательства | Требуемое повышение фактора свертывания крови VIII (% или МЕ/дл) | Частота введения доз (часы) / продолжительность терапии (дни) |
| Кровотечение | ||
| Ранняя фаза гемартроза, внутримышечное кровотечение или кровотечение в полости рта | 20-40 | Повторять инъекции каждые 12-24 часа, минимум в течение 1 суток до остановки кровотечения или купирования болевого синдрома. |
| Более выраженный гемартроз, внутримышечное кровотечение или гематома | 30-60 | Повторять инъекции каждые 12-24 часа в течение 3-4 дней или более до исчезновения болевого синдрома или острого нарушения функции. |
| Кровотечения, представляющие угрозу для жизни | 60-100 | Повторять инъекции каждые 8-24 часа до устранения угрозы для жизни. |
| Хирургическое вмешательство | ||
| Малое хирургическое вмешательство, включая неосложненное удаление зуба | 30-60 | Вводить каждые 24 часа, не менее 1 суток, до заживления раны. |
| Обширное хирургическое вмешательство | 80-100 (до и после операции) | Повторять инъекции каждые 8-24 часа до соответствующего заживления раны, затем еще не менее 7 дней для поддержания активности фактора свертывания крови VIII на уровне 30-60% (МЕ/дл). |
Профилактика
Рекомендованный начальный режим составляет от 20 до 50 МЕ/кг препарата Афстила® 2-3 раза в неделю. Режим можно корректировать в зависимости от клинического ответа пациента.
Дети
Дети в возрасте до 12 лет
Рекомендованный начальный режим у детей (в возрасте от 0 до <12 лет) составляет от 30 до 50 МЕ/кг препарата Афстила® 2-3 раза в неделю. У детей в возрасте <12 лет может потребоваться более частое введение или более высокие дозы с целью восполнения высокого клиренса, характерного для данной возрастной группы.
Дети старше 12 лет
Для подростков старше 12 лет рекомендации по дозированию аналогичны рекомендациям для взрослых пациентов
Пожилые
Клинические исследования препарата Афстила® не проводились у пациентов в возрасте старше 65 лет.
Инструкция по приготовлению раствора для внутривенного введения:
Общие инструкции
- Восстановленный раствор должен быть прозрачным или слегка опалесцирующим, бесцветным или желтоватым. После фильтрования/извлечения (см. ниже) и перед введением восстановленный раствор препарата следует проверить визуально на наличие механических включений и изменение цвета.
- Не использовать восстановленный раствор, в случае его помутнения или изменения цвета, а также, если в растворе наблюдаются механические включения.
- Приготовление восстановленного раствора препарата следует проводить в асептических условиях.
- Неиспользованный раствор или отходы следует утилизировать в соответствии с местными требованиями.
Приготовление восстановленного раствора
Довести растворитель до комнатной температуры. Убедиться, что крышки типа «флип-офф» с флаконов с растворителем и препаратом удалены, пробки обработаны антисептическим раствором и высушены до открытия устройства для добавления растворителя Mix2Vial.
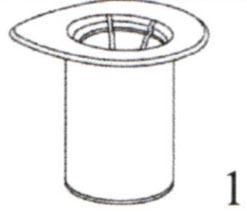 Снимите крышку с упаковки устройства для добавления растворителя со встроенным фильтром Mix2Vial. Не вынимайте устройство для добавления растворителя со встроенным фильтром Mix2Vial из блистерной упаковки!
Снимите крышку с упаковки устройства для добавления растворителя со встроенным фильтром Mix2Vial. Не вынимайте устройство для добавления растворителя со встроенным фильтром Mix2Vial из блистерной упаковки! 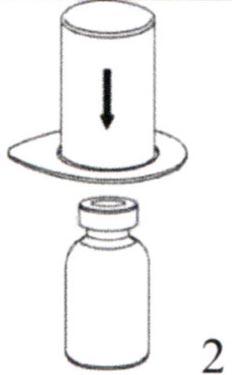 Поместите флакон с растворителем на ровную чистую поверхность, крепко удерживая его. Возьмите устройство Mix2Vial вместе с блистерной упаковкой и острым стержнем синей части устройства проткните пробку флакона с растворителем, надавливая вертикально вниз
Поместите флакон с растворителем на ровную чистую поверхность, крепко удерживая его. Возьмите устройство Mix2Vial вместе с блистерной упаковкой и острым стержнем синей части устройства проткните пробку флакона с растворителем, надавливая вертикально вниз
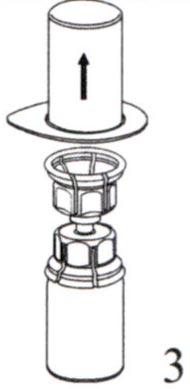 Аккуратно придерживая за край блистерной упаковки устройства Mix2Vial, снимите блистерную упаковку по направлению вертикально вверх. Убедитесь, что вы удалили только блистерную упаковку, а не само устройство Mix2Vial.
Аккуратно придерживая за край блистерной упаковки устройства Mix2Vial, снимите блистерную упаковку по направлению вертикально вверх. Убедитесь, что вы удалили только блистерную упаковку, а не само устройство Mix2Vial. 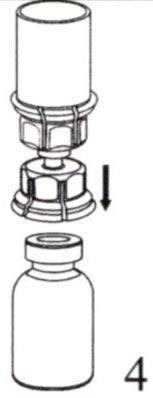 Поместите на ровную поверхность флакон с препаратом, и переверните над ним флакон с растворителем вместе с прикрепленным к нему устройством Mix2Vial, затем стержнем прозрачной части устройства для добавления растворителя проткните пробку флакона с препаратом, надавливая вертикально вниз. Растворитель автоматически переместится во флакон с препаратом.
Поместите на ровную поверхность флакон с препаратом, и переверните над ним флакон с растворителем вместе с прикрепленным к нему устройством Mix2Vial, затем стержнем прозрачной части устройства для добавления растворителя проткните пробку флакона с препаратом, надавливая вертикально вниз. Растворитель автоматически переместится во флакон с препаратом. 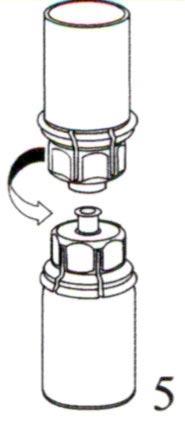 Одной рукой обхватите устройство для добавления растворителя Mix2Vial со стороны флакона с препаратом, другой – со стороны флакона с растворителем и, аккуратно развинтите устройство на две части, избегая сильного пенообразования при растворении лиофилизата. Флакон из под растворителя с синей частью присоединенного устройства для добавления растворителя Mix2Vial следует утилизировать.
Одной рукой обхватите устройство для добавления растворителя Mix2Vial со стороны флакона с препаратом, другой – со стороны флакона с растворителем и, аккуратно развинтите устройство на две части, избегая сильного пенообразования при растворении лиофилизата. Флакон из под растворителя с синей частью присоединенного устройства для добавления растворителя Mix2Vial следует утилизировать. 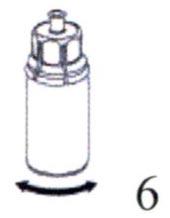 Аккуратно покрутите флакон с препаратом с присоединенным прозрачным устройством до полного растворения препарата. Не встряхивайте флакон.
Аккуратно покрутите флакон с препаратом с присоединенным прозрачным устройством до полного растворения препарата. Не встряхивайте флакон. 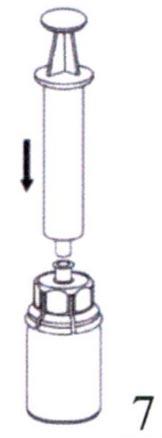 Наберите воздух в пустой стерильный шприц и, удерживая флакон с препаратом в вертикальном положении, присоедините шприц к наконечнику Люэра на устройстве Mix2Vial. Введите воздух во флакон с препаратом.
Наберите воздух в пустой стерильный шприц и, удерживая флакон с препаратом в вертикальном положении, присоедините шприц к наконечнику Люэра на устройстве Mix2Vial. Введите воздух во флакон с препаратом.
Забор и введение препарата
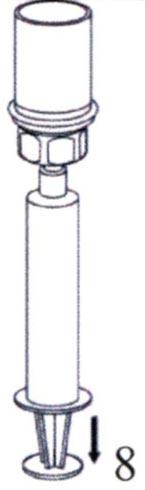 Нажимая на поршень шприца, переверните флакон вместе со шприцем. Плавно оттягивая поршень шприца, наберите в него восстановленный раствор.
Нажимая на поршень шприца, переверните флакон вместе со шприцем. Плавно оттягивая поршень шприца, наберите в него восстановленный раствор. 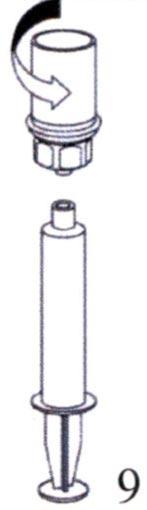 После того, как восстановленный раствор набран в шприц, обхватите цилиндр шприца (удерживая шприц вниз поршнем) и отсоедините прозрачное устройство Mix2Vial от шприца, поворачивая его против часовой стрелки.
После того, как восстановленный раствор набран в шприц, обхватите цилиндр шприца (удерживая шприц вниз поршнем) и отсоедините прозрачное устройство Mix2Vial от шприца, поворачивая его против часовой стрелки.
Для приготовления и введения препарата Афстила® следует использовать только прилагаемые в комплекте растворитель и устройство для добавления растворителя Mix2Vial, так как в связи с адсорбцией фактора свертывания крови VIII на внутренней поверхности некоторых устройств для введения лечение может оказаться неэффективным.
При проведении инъекции не допускать попадания крови в шприц с препаратом, так как это может вызвать коагуляцию крови в шприце и попадания сгустков в кровоток пациента.
Восстановленный раствор препарата Афстила® дальнейшему разведению не подлежит.
Восстановленный раствор препарата следует вводить в виде отдельной инъекции/инфузии внутривенно медленно. Приготовление восстановленного раствора проводят непосредственно перед применением.
Побочное действие:
Краткий обзор профиля безопасности
Гиперчувствительность или аллергические реакции (которые могут включать ангионевротический отек, жжение и зуд в месте введения препарата, озноб, покраснение, генерализованную крапивницу, локализованную крапивницу, головную боль, гипотензию, летаргию, тошноту, беспокойство, тахикардию, чувство стеснения в груди, звон в ушах, рвоту, свистящее дыхание) наблюдались редко при применении препаратов фактора свертывания крови VIII, и могут в некоторых случаях прогрессировать с развитием тяжелой анафилаксии (включая анафилактический шок).
Образование нейтрализующих антител (ингибиторов) может развиться у пациентов с гемофилией А, получающих фактор свертывания крови VIII, в том числе препарат Афстила®. При появлении ингибиторов на фоне лечения отмечается развитие недостаточного клинического ответа. В таких случаях рекомендуется обращаться в специализированный центр лечения гемофилии.
Табличный перечень нежелательных реакций
В таблице ниже представлены нежелательные реакции с частотой возникновения согласно классификации органов и систем-органов MedDRA. Частота развития, приведенная в таблице ниже, была установлена в завершенных клинических исследованиях у пациентов, ранее получавших лечение при тяжелой гемофилии А.
Частота указана по следующим категориям: очень часто (≥1/10), часто (≥1/100 до <1/10); нечасто (≥1/1000 до <1/100); редко (≥1/10000 до <1/1000) очень редко (<1/10000), неизвестно (частота не может быть оценена на основании имеющихся данных).
Таблица 4. Нежелательные реакции
| Классификация в соответствии с поражением органов и систем органов (MedDRA) | Нежелательные реакции | Частота встречаемости |
| Нарушения со стороны крови и лимфатической системы | Образование ингибиторов фактора VIII | нечасто (РЛП)* |
| Нарушения со стороны иммунной системы | Гиперчувствительность | часто |
| Нарушения со стороны нервной системы | Головокружение, парестезия | часто |
| Нарушения со стороны кожи и подкожных тканей | Сыпь | часто |
| Эритема, зуд | нечасто | |
| Общие расстройства и нарушения в месте введения | Повышение температуры тела | часто |
| Боль в месте инъекции, озноб, чувство жара | нечасто |
* Частота приведена в соответствии с клиническими исследованиями всех препаратов фактора свертывания крови VIII, включающих пациентов с тяжелой гемофилией А. РЛП = пациенты, ранее получавшие лечение.
Применение у детей
Различия в нежелательных реакциях, обусловленные возрастом, у пациентов детского возраста и взрослых пациентов отсутствовали.
Система сообщений о подозреваемых нежелательных реакциях
Необходимо сообщать о подозреваемых нежелательных реакциях после регистрации лекарственного препарата. Это позволяет проводить мониторинг соотношения польза/риск при применении лекарственного препарата. Специалистов здравоохранения просят сообщать о любых подозреваемых нежелательных реакциях.
Передозировка:
В завершенном клиническом исследовании пациент, получивший дозу, вдвое превышающую назначенную дозу препарата Афстила® (111 МЕ/кг), испытывал головокружение, чувство жара и зуд, расцененные как «не связанные с препаратом» Афстила®, а более вероятной причиной называли сопутствующее применение анальгетика. Другие случаи передозировки не описаны.
Взаимодействие с другими лекарственными средствами:
Нет данных о лекарственном взаимодействии препарата Афстила® с другими лекарственными средствами.
Фармацевтическая несовместимость
Для приготовления и введения препарата Афстила® следует использовать только прилагаемые в комплекте растворитель и устройство для добавления растворителя Mix2Vial. Не следует смешивать препарат Афстила® с другими лекарственными средствами в одном шприце или инфузионной системе.
Особые указания:
Для приготовления и введения препарата Афстила® следует использовать только прилагаемые в комплекте растворитель и устройство для разведения. При проведении инъекции не допускать попадания крови в шприц с препаратом, так как это может вызвать коагуляцию крови в шприце и попадания сгустков в кровоток пациента.
Гиперчувствительность
При применении препарата Афстила® возможно развитие аллергических реакций гиперчувствительности немедленного типа. При появлении симптомов гиперчувствительности применение препарата следует немедленно прекратить и обратиться за медицинской помощью.
Пациентов необходимо информировать о ранних симптомах реакций гиперчувствительности, таких как локализованная крапивница, генерализованная крапивница, чувство стеснения в груди, свистящее дыхание, гипотония и анафилаксия.
У пациентов с предшествующими реакциями гиперчувствительности следует провести соответствующую премедикацию.
В случае развития шока должна быть проведена стандартная противошоковая терапия.
Ингибиторы фактора свертывания крови VIII
У пациентов, получающих терапию препаратом Афстила®, необходимо проводить мониторинг образования нейтрализующих антител (ингибиторов) к фактору свертывания крови VIII, используя лабораторные и клинические методы. Обычно эти ингибиторы представляют собой иммуноглобулины IgG, действие которых направлено против прокоагулянтной активности фактора свертывания крови VIII, которые количественно определяют в единицах Бетезда (БЕ) на мл плазмы крови, используя модифицированный анализ. Риск образования ингибиторов коррелирует с тяжестью заболевания, также, как и экспозицией фактора свертывания крови VIII и наиболее высок в течение первых 20 дней введения. В редких случаях возможно образование ингибиторов после первых 100 дней введения препарата.
Также следует контролировать появление ингибиторов при переводе пациентов с одного препарата фактора свертывания крови VIII на другой, так как наблюдались случаи повторного образования ингибиторов (в низком титре) у пациентов, получивших терапию препаратами фактора свертывания крови VIII в течение более 100 дней. В связи с этим рекомендовано тщательное наблюдение за всеми пациентами в отношении образования ингибиторов после любой смены препарата.
Клиническая значимость образования ингибиторов зависит от титра ингибитора: низкий титр ингибиторов, который обнаруживается в течение короткого периода или присутствует постоянно, связан с меньшим риском недостаточного клинического ответа, в сравнении с высоким титром ингибиторов.
В целом следует проводить тщательный мониторинг в отношении всех пациентов, получающих терапию препаратами фактора свертывания крови VIII, в отношении образования ингибиторов посредством проведения соответствующих клинических наблюдений и лабораторных исследований. Если не удалось достичь ожидаемого уровня активности фактора свертывания крови VIII, или если кровотечение не остановилось после введения соответствующей дозы препарата, следует провести обследование пациента на наличие ингибиторов фактора свертывания крови VIII. У пациентов с высоким содержанием ингибиторов терапия фактором свертывания крови VIII может быть неэффективной, и в таких случаях следует рассмотреть вопрос о других вариантах лечения. Терапию таких пациентов должны проводить врачи, имеющие опыт лечения пациентов с гемофилией и ингибиторами фактора свертывания крови VIII.
Контроль лабораторных показателей
Если для определения активности фактора свертывания крови VIII у пациента используется одноступенчатый коагуляционный тест, его результат следует умножить на коэффициент пересчета 2. Для определения активности фактора свертывания крови VIII у пациента настоятельно рекомендуется использовать хромогенный анализ (см. раздел «Способ применения и дозы»).
Сердечно-сосудистые заболевания
У пациентов с имеющимися сердечно-сосудистыми факторами риска применение препаратов фактора свертывания крови VIII может повышать риск развития сердечнососудистых осложнений.
Осложнения, связанные с катетеризацией
При необходимости центрального венозного доступа (ЦВД) следует учитывать риск развития осложнений, связанных с ЦВД, включая местные инфекции, бактериемию и тромбоз в месте катетеризации.
Натрий
После восстановления препарат Афстила® содержит до 7 мг (0,3 ммоль) натрия в 1 мл раствора. Пациентам, соблюдающим диету с ограничением потребления натрия, следует это учитывать.
Документация при терапии препаратом
Настоятельно рекомендуется каждый раз при применении препарата Афстила® записывать наименование и номер серии препарата, чтобы обеспечить возможность отслеживания связи между пациентом и использованной серией лекарственного препарата.
Пациенты детского возраста
Перечисленные особые указания и меры предосторожности применимы как у взрослых, так и у детей.
Влияние препарата на способность управлять транспортными средствами или работать с механизмами:
Препарат Афстила® не оказывает влияния на способность управлять транспортными средствами и механизмами. Однако некоторые нежелательные реакции, связанные с действием препарата, могут оказывать влияние на концентрацию внимания и быстроту психомоторных реакций (см. раздел «Побочное действие»). Пациентам с подобными нарушениями следует воздержаться от управления транспортом, механизмами.
Форма выпуска:
Лиофилизат для приготовления раствора для внутривенного введения, 250 ME, 500 ME, 1000 ME, 1500 ME, 2000 ME, 2500 ME или 3000 ME, в комплекте с растворителем (вода для инъекций).
По 250 ME, 500 ME или 1000 ME во флакон вместимостью 6 мл или 1500 ME, 2000 ME, 2500 ME или 3000 ME во флакон вместимостью 10 мл из прозрачного бесцветного стекла типа I, укупоренный пробкой из бромбутилового каучука и обкатанный алюминиевым колпачком с пластиковой крышкой типа «флип-офф».
По 2,5 или 5,0 мл воды для инъекций во флакон прозрачного бесцветного стекла типа I, укупоренный пробкой из бромбутилового или хлорбутилового каучука и обкатанный алюминиевым колпачком с пластиковой крышкой типа «флип-офф».
Устройство для добавления растворителя со встроенным фильтром 15 мкм (Mix2Vial) в блистер из полиэтилентерефталата/бумаги, ламинированной полиэтиленом.
По 1 флакону с лиофилизатом, 1 флакону с растворителем, 1 устройству для добавления растворителя со встроенным фильтром, и 1 картонной пачке с комплектом для внутривенного введения препарата (1 одноразовым шприцем объемом 5 или 10 мл, 1 игле-бабочке, 2 дезинфицирующими салфетками в индивидуальных герметичных упаковках и 1 нестерильным лейкопластырем) с контролем первого вскрытия и инструкцией по медицинскому применению в картонную пачку с контролем первого вскрытия.
Условия хранения:
Хранить при температуре от 2 до 8 °С. Не замораживать.
Допускается единовременное хранение при комнатной температуре до 25 °С в течение 3 месяцев в пределах срока годности, указанного на этикетке флакона и на картонной пачке.
После изъятия препарата из холодильника не допускается его повторное хранение в холодильнике. Дату изъятия препарата из холодильника для начала хранения при комнатной температуре следует указать на картонной пачке.
Хранить флакон с препаратом в оригинальной упаковке для защиты от света.
Хранить в недоступном для детей месте.
Срок годности:
3 года.
Не применять по истечении срока годности.
Условия отпуска:
Отпускают по рецепту.
Производитель:
СиЭсЭл Беринг ГмбХ, Эмиль-фон-Беринг-Штрассе 76, 35041 Марбург, Германия
CSL Behring GmbH, Emil-von-Behring-StraBe 76, 35041 Marburg, Germany
Владелец РУ:
СиЭсЭл Беринг ГмбХ, Эмиль-фон-Беринг-Штрассе, 35041, Марбург, Германия
CSL Behring GmbH, Emil-von-Behring-StraBe 76, 35041 Marburg, Germany
Организация, уполномоченная на принятие претензий:
Филиал Общества с ограниченной ответственностью «Си Эс Эл Беринг Биотэрапис ГмбХ»
125167, г. Москва, Ленинградский проспект, д. 39, стр. 80
Комментарии
ПРАКТИКА ПЕДИАТРА



