
Амилоидная нефропатия у ребенка с периодической болезнью
Статьи
![]()
ЖУРНАЛ "ПРАКТИКА ПЕДИАТРА"
Опубликовано в журнале:
Практика педиатра, февраль, 2015
М.Е. Аксенова, к. м. н., В.В. Длин, д. м. н., профессор, О.Ю. Турпитко, к. м. н, В.В. Невструева, вед. н. с., Обособленное структурное подразделение «Научно-исследовательский клинический институт педиатрии» ГБОУ ВПО РНИМУ им. Н. И. Пирогова Минздрава РФ, г. Москва Ключевые слова: нефротический синдром, амилоидоз, периодическая болезнь, дети.
Key words: nephrotic syndrome, amyloidosis, familial mediterranean fever, children.
Вторичный нефротический синдром в детском возрасте чаще бывает обусловлен системными болезнями соединительной ткани (системная красная волчанка, васкулиты), инфекциями (вирусные гепатиты, малярия, сифилис, ВИЧ-инфекция) или отмечается в рамках генетических синдромов, реже – осложняет сахарный диабет, неопластические процессы, болезни накопления, лекарственная терапия.
Амилоидоз почек – очень редкая причина нефротического синдрома в детском возрасте. У детей амилоидная нефропатия чаще всего развивается на фоне хронических воспалительных заболеваний (остеомиелит, ревматоидный артрит) и периодической болезни [1], при которой она встречается в 40–60% случаев [1, 2]. Прогноз больных с амилоидозом почек, как и при других вариантах вторичного нефротического синдрома, зависит от своевременного назначения специфической терапии.
Под нашим наблюдением находился ребенок с нефротическим синдромом, развившимся на фоне амилоидоза почек, осложнившего периодическую болезнь.
Мартин Х., 10.03.1990, армянин, поступил в отделение нефрологии МНИИ педиатрии и детской хирургии МЗ РФ мае 2000 года в крайне тяжелом состоянии с диагнозом «гормонорезистентный нефротический синдром». Отец ребенка страдает мочекаменной болезнью, тетя по линии отца имеет порок развития мочевой системы в виде единственной почки, у некоторых родственников по линии матери отмечаются рецидивирующие боли в животе неуточненного генеза. Больной родился от 3-й, протекавшей с легким токсикозом беременности, 2-х физиологических родов. На 1-м году жизни часто болел отитами, в 2,5 года перенес ожог левой шейно-лицевой области, после которого остался грубый келоидный рубец. С 3 лет у мальчика появились рецидивирующие боли в животе, сопровождавшиеся субфебрильной температурой. Болевой синдром продолжался 3–4 дня и уменьшался на фоне приема аспирина. В 5 лет при обследовании по поводу слабости, болей в коленных и голеностопных суставах у ребенка были выявлены увеличение печени, ускорение СОЭ до 45 мм/ч, анализы мочи – без патологии. Больной наблюдался с диагнозом «ревматизм, рецидивирующее течение», не лечился. В 9 лет мальчик был оперирован по поводу аппендицита. В постоперационном периоде у ребенка впервые были выявлены отеки голеней, протеинурия до 3 г/сут, анемия, гипо- и диспротеинемия с повышением уровня гамма глобулинов до 34%, повышение липидов и холестерина крови. Больному был поставлен диагноз «гломерулонефрит, нефротическая форма». Изолированная 4-недельная терапия преднизолоном (2 мг/кг/сут) и последующая сочетанная терапия лейкераном (0,2 мг/кг/сут) и стероидами (60 мг/сут) в течение 8 недель эффекта не дали. В тяжелом состоянии мальчик был госпитализирован в отделение нефрологии МНИИ педиатрии и ДХ МЗ РФ.
При осмотре у больного отмечались: анасарка, бледность, дистрофия кожи и ее придатков, обширный келоидный рубец левой шейно-лицевой области с деформацией ушной раковины, единичные пятнисто-папулезные и геморрагические кожные элементы на голенях, глоссит, ангулярный стоматит, гепатоспленомегалия. Выявлялись умеренная синусовая тахикардия, повышение артериального давления до 130/90 мм рт. ст. Осложнения предшествующей стероидной терапии проявлялись ожирением, гипостатурой (рост ребенка соответствовал 10-й возрастной перцентили), выраженным остеопорозом, рецидивирующими паронихиями. В крови отмечались лейкоцитоз со сдвигом лейкоцитарной формулы влево (лейкоциты 16,8 тыс., п/я 10%, метамиелоциты 2%, миелоциты 1%), анемия (эр. 3,79 млн, Hb 106 г/л), тромбоцитоз до 500 тыс., ускорение СОЭ до 57 мм/ч, выраженная гиперкоагуляция. Общий белок крови составлял 37 г/л, альбумины – 11,5 г/л, глобулины альфа-1 – 4,5%, альфа-2 – 43%, бета – 10%, гамма – 11%, холестерин – 9,42 ммоль/л, липиды – 16,3 ммоль/л. Уровень IgG крови был снижен до 0,1 г/л, IgA – до 0,25 г/л. Протеинурия умеренной селективности превышала 8 г/сут, в осадке мочи определялись единичные эритроциты, гиалиновые цилиндры. Отмечалось снижение клиренса эндогенного креатинина до 65 мл/мин при нормальном уровне креатинина и мочевины крови (0,066 ммоль/л и 4,4 ммоль/л соответственно), снижение экскреции титруемых кислот с мочой; концентрационная функция почек была сохранна. При УЗИ выявлялось увеличение почек в объеме в 2,5 раза от возрастной нормы, значительное повышение эхогенности паренхимы почек.
Учитывая наличие гормонорезистентного нефротического синдрома, полиорганный характер поражения, дифференциальный диагноз проводился с вторичным гломерулонефритом на фоне системных заболеваний соединительной ткани и васкулитов. Геморрагический васкулит был исключен на основе отсутствия у больного типичных кожного и суставного синдромов. Отсутствие у мальчика подкожных узелков, злокачественной артериальной гипертензии позволило исключить узелковый периартериит. Отрицательные результаты исследования крови на маркеры системной патологии (антитела к нативной и денатурированной ДНК, антитела к антигенам кардиомиоцитов, эндотелия, гладких мышц, антинуклеарный фактор, ревматоидный фактор, LE-клетки), проведенные у ребенка на фоне приема преднизолона, позволили предположительно исключить системную красную волчанку и другие аутоиммунные заболевания.
Учитывая этнический фактор, наличие у родственников рецидивирующих болей в животе, наличие у больного рецидивирующего абдоминального синдрома, сопровождающегося повышением температуры, суставного синдрома, гормонорезистентного нефротического синдрома, гипергаммаглобулинемии в анамнезе, впервые была заподозрена периодическая болезнь, осложненная амилоидозом почек.
Доза преднизолона была постепенно снижена до 25 мг/сут, больной получал симптоматическую терапию: 20% р-р альбумина, верошпирон 6 мг/кг/сут, гепарин 250 Ед/кг/сут, капотен 1 мг/кг/сут, глюконат кальция 1,5 г/сут, оксидевит 50 мкг/сут.
В период подготовки к нефробиопсии у ребенка развился компрессионный перелом грудного отдела позвоночника (06.2000), что потребовало назначения корсета, курсов остеогенона и электрофореза с кальцием на область позвоночного столба. В августе 2000 года у пациента появились боли в мышцах верхних конечностей, плечевого пояса, шеи, интенсивность которых нарастала при движении и пальпации, подкожно определялись болезненные узелки до 0,5 см в диаметре. Мышечный синдром регрессировал самостоятельно. Через несколько дней у больного развился тромбоз глубоких вен правой нижней конечности, доза гепарина была увеличена, были назначены аспирин, местные аппликации с гепариновой мазью. В конце августа у ребенка появились острые боли в животе, с подозрением на острую хирургическую патологию мальчик был переведен в хирургическое отделение ДКБ № 9. Динамическое наблюдение за ребенком, результаты обследования позволили исключить хирургические болезни. 08.09.2000 больному была проведена полуоткрытая биопсия левой почки. После оперативного вмешательства состояние мальчика резко ухудшилось: наросли отечный синдром вплоть до развития анасарки, артериальное давление до 140–150/85–100 мм рт. ст., появилась тахикардия до 94 уд/мин. По данным лабораторного обследования отмечалось нарастание анемии (Нв 79 г/л), СОЭ до 75 мм/ч, признаков нефротического синдрома, гиперкоагуляции. Появились электролитные нарушения (калий крови – 3,4 ммоль/л, натрий – 132 ммоль/л), повышение мочевины крови до 13,3 ммоль/л, снижение процессов реполяризации в миокарде желудочков по данным ЭКГ. Уровень креатинина крови оставался нормальным (0,06 ммоль/л). 19.09.2000 больной был переведен в отделение интенсивной терапии, где наряду с симптоматическим лечением (верошпирон 6,5 мг/кг/сут, курантил 3 мг/кг/сут, гепарин 250 Ед/кг/сут, капотен 1,5 мг/кг/сут, атенолол 0,2 мг/кг/сут, глюконат кальция 2 г/сут, оксидевит 50 мкг/сут, остеогенон), получал в/в инфузии онкотических растворов (20% р-р альбумина, инфукол), петлевые диуретики, антибиотики (фортум 2 г/ сут). Несмотря на проводимую терапию, у ребенка развились сердечно-сосудистая недостаточность, отек мозга. 23.09.2000 больной умер.
Морфобиоптическое исследование почек выявило характерные признаки амилоидной нефропатии (рис.). В нефробиоптатате при световой микроскопии выявлялось диффузное расположение амилоида практически во всех гломерулах, стенках мелких и средних сосудов почки, интерстиции, гиалино-капельная и зернистая дистрофия канальцев, незначительная лимфо-макрофагальная инфильтрация интерстициальной ткани и умеренный склероз коркового и мозгового вещества почки. Иммуногистохимическое исследование препаратов почечной ткани, проведенное к. м. н. Проскурниковой Е.П. (кафедра патологической анатомии ММА им. И.М. Сеченова), выявило отложение амилоида АА-типа.
По данным патологоанатомического исследования, проведенного зав. отд. патологической анатомии ДКБ № 7 к. м. н. Катасоновой Л.П., причиной смерти ребенка послужил отек мозга с вклинением затылочных долей в большое затылочное отверстие, развившийся на фоне периодической болезни. При патологоанатомическом вскрытии у больного были выявлены признаки периодической болезни в виде формирования рыхлых спаек между париетальной и висцеральной плеврой, сердечной сорочкой и эпикардом, кишечными петлями, париетальной и висцеральной брюшиной, амилоидоз почек, печени, селезенки, бифуркационных лимфоузлов.
Амилоидоз – групповое понятие, объединяющее заболевания, отличительным признаком которых является внеклеточное отложение в тканях специфического нерастворимого фибриллярного белка – амилоида. Современная классификация амилоидоза основана на специфичности основного белка, формирующего амилоид. С клинической точки зрения целесообразно выделять системные и локальные, наследственные и приобретенные формы амилоидоза. Частота амилоидоза у взрослых составляет около 1,5%, при этом поражение почек развивается более чем у трети больных [2–4].
Периодическая болезнь – заболевание с аутосомно-рецессивным (MIM#249100), гораздо реже аутосомно-доминантным (MIM#134610) [5, 6] типами наследования с выраженной этнической принадлежностью: чаще болеют арабы, евреи, турки, армяне. Ген болезни MEFV (mediterranean fever) картирован на коротком плече 16-й хромосомы (16p13.3) и кодирует белок пирин (маренострим), контролирующий хемотаксис нейтрофилов [7]. Мутация гена приводит к развитию неконтролируемой воспалительной реакции. Ген MEFV экспрессируется главным образом в нейтрофилах, эозинофилах, моноцитах, в меньшем количестве – в дендритных клетках, фибробластах кожи, брюшины и синовиальной оболочки [6, 7]. Этим объясняются основные клинические проявления периодической болезни: лихорадка, полисерозиты, поражение кожи. Экспрессия гена регулируется провоспалительными факторами: интерлейкином-1-бета, интерфероном-гамма, фактором некроза опухоли-альфа.
Мутации MEFV выявляются в разных популяциях с частотой 0,02–0,22% [6, 8], клинические симптомы заболевания развиваются только у 1 из 9 носителей мутировавшего гена болезни [8]. В настоящее время идентифицировано более 60 мутаций гена MEFV (fmf.igh.cnrs.fr/ISSAID/infevers). Основной процент приходится на мутации M680I, M694V, M694I, V726A, E148Q, каждая из которых превалирует в определенной этнической группе [8–10]. Практически все эти мутации определяют структурные изменения домена B30.2 пирина, связывающего различные патологические протеины с последующей активацией аутовоспалительной реакции.
Мутация M694V (на ее долю приходится 60–80% всех мутаций MEFV), особенно в гомозиготном состоянии, коррелирует с тяжелым фенотипом болезни и развитием амилоидоза [7]. Кроме того, показано, что несколько генов оказывают модифицирующее влияние на течение периодической болезни, увеличивая риск развития амилоидоза: ген SAA1, определяющий альфа/альфа-фенотип амилоида А, и MICA ген (Major Histocompatibility Complex, MCH class-I-chain-related type A) [6].
Клинические признаки периодической болезни чаще появляются на 5–6-м году жизни [1, 8, 9], более чем у 95% пациентов болезнь дебютирует до 20 лет в виде лихорадки, рецидивирующих абдоминальных болей, которые отмечаются более чем у 80% больных, поражения суставов (30–50% больных), кожной сыпи, чаще по типу узловой эритемы (15–30%), гепатоспленомегалии [2, 8, 9]. Характерным и диагностически важным признаком является стереотипность приступов. В период между приступами болезненные проявления отсутствуют. Длительность приступов составляет от 12 часов до 3 суток, межприступный период продолжается от нескольких дней до нескольких месяцев и значительно варьирует у одного и того же больного. В 1997 году были предложены клинические критерии постановки диагноза «периодическая болезнь», однако из-за низкой специфичности они так и не получили широкого применения в клинике.
Периодическая болезнь как хроническое аутовоспалительное заболевание приводит к развитию вторичного АА-амилоидоза приблизительно у трети больных. При этом у половины пациентов развивается амилоидная нефропатия – основное и самое грозное осложнение периодической болезни, определяющее дальнейший прогноз больных.
Поражение почек отмечается, как правило, через 5–8 лет от манифестации периодической болезни и проходит в своем развитии 3 стадии: протеинурическую (пренефротическую), нефротическую и азотемическую [2, 4]. Протеинурия носит, как правило, изолированный характер. Ее появление связано с отложением амилоида между подоцитами и капиллярной базальной мембраной почечных клубочков. Чем длиннее протеинурическая стадия болезни, тем лучше прогноз пациентов: 5-летняя почечная выживаемость составляет 74%, а 10-летняя – 56% при длительности изолированной протеинурии до 3 лет, по сравнению со 100% и 83% соответственно – при пролонгированной пренефротической стадии амилоидоза почек [2]. Нефротический синдром развивается у 80% больных с почечным амилоидозом через 3–13 лет от появления изолированной протеинурии [2]. При этом отеки появляются достаточно рано, приобретают распространенный и упорный характер, оставаясь значительными и в период терминальной почечной недостаточности. Наряду с массивной протеинурией в развитии отечного синдрома при амилоидозе играют роль повышенный катаболизм белка в организме больного, уменьшение всасывания и потеря белка через желудочнокишечный тракт, нарушение белково-синтетической функции печени. При естественном течении болезни через 5 лет от развития нефротического синдрома в живых остается менее половины больных, ХПН необратимо развивается через 2–13 лет от появления клинических признаков амилоидной нефропатии [1–4]. Реже проявлениями амилоидоза почек могут быть тубулярная дисфункция и почечная недостаточность. Почечная недостаточность как начальное проявление амилоидной нефропатии отмечается у 12,5% больных [2, 7]. Ее развитие обусловлено преимущественным поражением почечных сосудов. Быстрое прогрессирование почечной недостаточности при амилоидозе почек может быть связано с тромбозом почечных вен и/или выраженной атрофией канальцев. Тубулоинтерстициальные нарушения с развитием интерстициального фиброза в большей степени, чем поражение гломерул, коррелируют с функциональными нарушениями почек, поэтому выявление канальцевой дисфункции имеет важное прогностическое значение [2, 4].
Реже при вторичном амилоидозе на фоне периодической болезни поражаются другие органы: печень (около 50% больных), селезенка (30–40%), кишечник (20%) и практически отсутствуют поражения сердца, респираторного тракта, периферической и вегетативно-нервной системы, поперечно-полосатой мускулатуры.
К лабораторным признакам, характерным для амилоидоза почек, относятся: повышение СОЭ более 50 мм/ч, которое выявляется у 3/4 пациентов, диспротеинемия с повышением уровня гамма глобулинов крови, выявляемая у 40% больных, и наличие SAA-белка – предшественника амилоида в крови [4, 12]. Показано, что скорость прогрессирования амилоидной нефропатии коррелирует с концентрацией SAA-протеина в крови. Для прогнозирования течения амилоидоза может также использоваться концентрация более распространенного в клинической практике маркера воспаления – С-реактивного белка [3].
Типичным для амилоидоза является гепатои нефромегалия при УЗИ. Обычно почки значительно увеличены в размерах, даже на стадии ХПН, с нарушенной дифференцировкой паренхимы на корковый и медуллярный слои, иногда (10% случаев) может отмечаться неровность контуров почек из-за очаговой атрофии коры вследствие артериолосклероза и/или окклюзии почечных сосудов амилоидными массами.
В клинической практике, особенно для контроля за степенью амилоидоза и эффективностью терапии, может использоваться сцинтиграфия с 123I-меченным сывороточным Р-компонентом (SPP), который специфически связывается с амилоидными депозитами любого типа.
Диагноз «амилоидоз» подтверждается морфологически. Информативным методом является исследование биоптата стенки прямой кишки (слизистого и подслизистого слоя), почек/печени, при которых амилоид выявляется в 50–70% и 90–100% случаев соответственно. При световой микроскопии амилоид имеет вид аморфных эозинофильных масс, а при окраске конго красным (розово-красное окрашивание) дает характерное зеленое свечение в поляризованном свете.
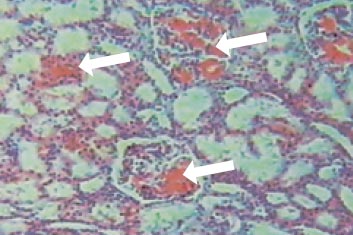
Фрагмент нефробиоптата больного М, 10 лет, амилоидоз почек. Световая микроскопия, увеличение в 110 раз, окраска конго рот. Конгломераты амилоида в гломерулах, в стенке артериол
Анализируя историю заболевания нашего больного, можно отметить, что у пациента периодическая болезнь манифестировала в 3 года с характерных фебрильных абдоминальных кризов, к которым позднее присоединились поражение суставов, гепатоспленомегалия, резистентный к стероидам и диуретикам нефротический синдром. При лабораторном обследовании у мальчика выявлялись выраженные ускорение СОЭ, гипергаммаглобулинемия, анемия при сохранной функции почек. В семье больного несколько родственников страдали периодическими болями в животе. Учитывая этническую принадлежность, все это еще на ранних этапах заболевания позволяло заподозрить у ребенка периодическую болезнь, осложненную амилоидозом почек, и могло способствовать назначению адекватной терапии.
Правильно и своевременно диагностированная периодическая болезнь позволяет не только назначить специфическое лечение, улучшить прогноз пациентов, но и избежать повторных неоправданных хирургических и медикаментозных вмешательств.
При лечении АА-амилоидоза в рамках периодической болезни препаратом выбора является колхицин (1,5–2 мг/сут), который блокирует образование амилоидускоряющего фактора, ингибирует синтез и секрецию SAA, влияя на хемотаксическую активность полиморфноядерных лейкоцитов крови [2, 6, 11–14]. Применение колхицина полностью предотвращает развитие симптомов периодической болезни у 65% больных, а у 30% приводит к регрессу клинической симптоматики. Эффективность колхицина в регрессе амилоидоза почек, обратном развитии нефротического синдрома дискутабельна. По данным Кочубей Л.Н., Виноградовой О.М. [2], положительный эффект терапии в виде уменьшения или исчезновения нефротического синдрома отмечался у 95% из 47 больных, получавших колхицин в течение 4 лет. При этом продолжительность жизни больных от момента выявления нефротического синдрома в результате применения колхицина возросла с 2,7 до 9,6 лет, от момента выявления признаков ХПН – с 1,6 до 5,5 лет. Исчезновение нефротического синдрома с сохранением изолированной протеинурии при более чем 3-летнем приеме колхицина отмечалось у 13 из 38 детей (q=0,34), при этом препарат не был эффективен у пациентов с уже сформировавшейся на момент начала терапии ХПН [15].
В настоящее время ведутся активные поиски новых методов терапии периодической болезни, так как 5–10% пациентов имеют резистентность к колхицину, еще 2–5% больных прекращают лечение в связи с непереносимостью/токсичностью препарата. Блокаторы рецепторов интерлейкина-1 (anakinra, rilonacept) [13, 14] и фактора некроза опухоли-альфа (infl iximab) [15] значительно уменьшают симптоматику заболевания и могут быть использованы у пациентов, не отвечающих на традиционное лечение, особенно при наличии клиники системного васкулита.
Для лечения вторичного АА-амилоидоза в практике применяется диметилсульфоксид (ДМСО), способствующий резорбции амилоидных отложений [2, 16]. Препарат применяется в дозе не менее 10 г/сут в течение 6 месяцев и более. К потенциально эффективным методам терапии АА-амилоидоза можно отнести eprodisate, препятствующий взаимодействию амилоидогенных протеинов с глюкозамингликанами и ингибирующий полимеризацию и отложение амилоидных фибрилл в тканях [15].
В лечении амилоидной нефропатии на стадии терминальной почечной недостаточности широко применяются методы заместительной терапии. По данным Российского регистра заместительной почечной терапии, амилоидоз почек является причиной тХПН у 0,76% пациентов, получавших заместительную почечную терапию в 2009 году [17].
Выживаемость больных с амилоидозом почек на гемодиализе не отличается от таковой у больных с другими системными заболеваниями (например, с сахарным диабетом) [2]. После трансплантации показатели выживаемости больного и трансплантата сопоставимы с данными показателями в группах пациентов с другой почечной патологией [2]. Реамилоидоз трансплантата развивается у 17% больных с периодической болезнью [2].
Таким образом, одной из причин развития вторичного нефротического синдрома у детей является редко встречающаяся в педиатрической практике амилоидная нефропатия. Амилоидоз почек в первую очередь должен быть заподозрен у больных с клиникой периодической болезни (сочетание лихорадки, рецидивирующих болей в животе, суставного синдрома у детей определенных этнических групп) и другими аутовоспалительными заболеваниями или у пациентов с длительно текущими аутоиммунными или гнойно-деструктивными болезнями. К особенностям нефротического синдрома при амилоидной нефропатии относятся: тяжелый, резистентный к терапии отечный синдром, значительное повышение СОЭ и белков острой фазы воспаления (C-реактивный белок, SAA-протеин), гипергаммаглобулинемия, анемия при сохранной функции почек. Прогноз заболевания серьезный и во многом зависит от своевременного назначения специфического лечения.
Список литературы:
- Tinaztepe K. Renal amyloidosis in childhood. An overwiew of the topic with 25 years experience // J. Turk. Pediatr., 1995; 37 (4): 357–73.
- Мухин Н.А., Серов В.В., Козловская Л.В. Амилоидоз почек. Нефрология: руководство для врачей под ред. И.Е. Тареевой. М.: Медицина, 2000: 546–556.
- Joss N., Boulton-Jones M. Amyloidosis. In: Davison A.M., Cameron J.S., Grunfeld J.P. et al. Oxford Textbook of Clinical Nephrology. Oxford: «Oxford Publications», 2008: 679–701.
- Рамеев В.В., Симонян А.Х., Саркисова И.А. и соавт. Амилоидоз и наследственные периодические аутовоспалительные заболевания // Клиницист, 2008; 2: 6–15.
- Шулутко Б.И. Болезни печени и почек. СПб санитарно-гигиенический мединститут, 1993: 373–384.
- Livnech A., Zemer D., Siegal B. Colchicine prevents kidney transplantant amyloidosis in familial mediterranean fever. Nephron, 1992; 60: 418–22.
- Aldea A., Campistol J.M, Arostegui J.I. et al. A severe autosomal-dominant periodic infl ammatory disorder with renal AA amyloidosis and colchicine resistance associated to the MEFV H478Y variant in a Spanish kindred: an unusual familial mediterranean fever phenotype or another MEFVassociated periodic infl ammatory disorder? // Am. J. Med. Genet. A., 2004; 124A (1): 67–73.
- Ozen S. New interest in an old disease: familial mediterranean fever // Clin. Exp. Rheumatol., 1999; 17 (6): 745–9.
- Medlej-Hashim M., Loiselet J., Lefranc G., Megarbane A. Familial Mediterranean Fever (FMF): from diagnosis to treatmen. Sante, 2004; 14 (4): 261–6.
- Ozcakar Z.B., Yuksel S., Ekim M. et al. Infl iximab therapy for familial mediterranean feverrelated amyloidosis: case series with long term follow-up // Clin. Rheumatol., 2012; 31 (8): 1267–71.
- Daniel L. Kastner. Advances in the understanding of familial mediterranean fever and possibilities for targeted therapy // Br. J. Haematol., 2009; 5: 467–478.
- Ben-Chetrit E. Familial mediterranean fever (FMF) and renal AA amyloidosis – phenotype-genotype correlation, treatment and prognosis // J. Nephrol., 2003; 3: 431–434.
- Breuning M.H., Bakker E. From gene to disease; marenostrine and familial mediterranean fever. Nederlands Tijdschrift Voor Geneeskude, 2000; 36: 1728–30.
- Soriano A., Verecchia E., Afeltra A. et al. IL1? biological treatment of familial Mediterranean fever. Clin. Rev. Allergy Immunol., 2013; 1: 16–24.
- Dember L.M., Hawkins P.N., Hazenberg B.P. et al. Eprodisate for the treatment of renal disease in AA amyloidosis // N. Engl. J. Med., 2007; 356: 2349–60.
- Tan S.Y., Pepys M.B., Hawkins P.N. Treatment of amyloidosis // Am. J. Kidney Dis., 1995; 26 (2): 267–285.
- Бикбов Б.Т., Томилина Н.А. Состояние заместительной терапии больных с хронической почечной недостаточностью в Российской Федерации в 1998–2009 гг. (отчет по данным Российского регистра заместительной почечной терапии) // Нефрология и диализ, 2011; 3: 150–264.



