Efficacy and safety of asenapine in a placebo- and haloperidol-controlled trial in patients with acute exacerbation of schizophrenia
Статьи Опубликовано в журнале:«JOURNAL OF CLINICAL PSYCHOPHARMACOLOGY»; Volume 30; Number 2; April; 2010; стр. 106-115.
John M. Kane, MD,* Michael Cohen, PhD, † Jun Zhao, PhD, † Larry Alphs, MD, PhD,‡ and John Panagides, PhD†
Abstract: Asenapine is approved by the Food and Drugs Administration in adults for acute treatment of schizophrenia or of manic or mixed episodes associated with bipolar i disorder with or without psychotic features. In a double-blind 6-week trial, 458 patients with acute schizophrenia were randomly assigned to fixed-dose treatment with asenapine at 5 mg twice daily (BID), asenapine at 10 mg BID, placebo, or haloperidol at 4 mg BiD (to verify assay sensitivity). With last observations carried forward (LOCF), mean Positive and Negative Syndrome Scale total score reductions from baseline to endpoint were significantly greater with asenapine at 5 mg BID ( — 16.2) and haloperidol ( — 15.4) than placebo (—10.7; both P <0.05); using mixed model for repeated measures (MMRM), changes at day 42 were significantly greater with asenapine at 5 and 10 mg BID (—21.3 and —19.4, respectively) and haloperidol (—20.0) than placebo (—14.6; all P <0.05). On the Positive and Negative Syndrome Scale positive subscale, all treatments were superior to placebo with LOCF and MMRM; asenapine at 5 mg BID was superior to placebo on the negative subscale with MMRM and on the general psychopathology subscale with LOCF and MMRM. Treatment-related adverse events (AEs) occurred in 44% and 52%, 57%, and 41% of the asenapine at 5 and 10 mg BID, haloperidol, and placebo groups, respectively. Extrapyramidal symptoms reported as AEs occurred in 15% and 18%, 34%, and 10% of the asenapine at 5 and 10 mg BID, haloperidol, and placebo groups, respectively. Across all groups, no more than 5% of patients had clinically significant weight change. Post hoc analyses indicated that efficacy was similar with asenapine and haloperidol; greater contrasts were seen in AEs, especially extrapyramidal symptoms.
Key Words: asenapine, efficacy, haloperidol, safety, schizophrenia
Schizophrenia is a chronic illness marked by varying symptoms, including the overt positive symptoms, the less salient negative symptoms, and some level of cognitive impairment. Currently available antipsychotic medications, both conventional and atypical agents, are effective for treating acute exacerbations of schizophrenia. As maintenance medications, these drugs are effective for positive symptoms1 but have a less consistent effect on negative symptoms.2 A principal advantage of the atypical antipsychotics is that they are less likely than conventional agents, such as haloperidol, to cause extrapyramidal symptoms (EPS) at recommended doses.3
Nevertheless, there is a very high incidence of discontinuation of prescribed medication among patients with schizophrenia.4 Although discontinuation of pharmacotherapy is often related to poor therapeutic response,5 other factors not related to drug treatment may also influence discontinuation rates (eg, current substance abuse or comorbid psychiatric illness, poor level of insight, and schizophrenia symptom severity) in clinical trials.6,8
Asenapine is indicated in the United States for the acute treatment of schizophrenia in adults and the acute treatment of manic or mixed episodes associated with bipolar I disorder with or without psychotic features in adults. It has potent antagonistic activity at dopaminergic, serotonergic, ?-adrenergic, and histaminic receptors but no appreciable affinity for muscarinic receptors.9 Sublingually administered asenapine is rapidly absorbed. Peak asenapine plasma concentrations of approximately 4 ng/mL are reached approximately 1 hour after sublingual dosing.10 The administration of a strong CYP1A2 inhibitor, such as fluvoxamine, or severe hepatic impairment has been reported to increase asenapine exposure11,12; exposure to asenapine is not altered in a clinically relevant manner by renal impairment or by mild to moderate hepatic impairment.11
The asenapine schizophrenia clinical trials program has compared asenapine to placebo, with different agents being used as active comparators (ie, olanzapine, risperidone, and haloperidol). The Hera studies encompass fixed-dose short-term trials in acute schizophrenia as well as flexible-dose longer term extension studies. In a published randomized, double-blind, placebo-and risperidone-controlled phase 2 trial enrolling 182 patients, fixed-dose asenapine (5 mg twice daily [BID]) was effective and well tolerated in the treatment of acute schizophrenia.13 The present study (Hera 041023: clinical trials registry number, NCT00156104) was a large-scale, phase 3, multicenter, global study that used 2 fixed dosages of asenapine to further assess the efficacy, tolerability, and safety ofasenapine in the treatment of schizophrenia. It compared asenapine to placebo while using haloperidol as an active control to assess assay sensitivity. Although second-generation antipsychotics are increasingly being used as first-line agents in the treatment of schizophrenia,14 haloperidol is still commonly used in view of its demonstrated efficacy and low cost.15
Methods
This randomized, double-blind, placebo-controlled trial was conducted at 43 centers (United States, 17 sites; Russia, 11 sites; India, 7 sites; Romania, 7 sites; Canada, 1 site) from June 2005 to September 2006. The protocol was approved by the independent ethics committee/institutional review board at each site, and the study was conducted in accordance with the Declaration of Helsinki and ICH Guidelines for Good Clinical Practice. Written informed consent was obtained from each patient. Subjects unable to provide consent could participate if a legally authorized representative provided consent and the subject affirmed his or her participation by signing an assent form.
Eligible patients were aged 18 years or older and had a Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision, diagnosis of schizophrenia with an acute exacerbation of psychotic symptoms at study enrollment. Other principal inclusion criteria were a Positive and Negative Syndrome Scale16 (PANSS) total score of 60 or higher, with scores of 4 or higher on at least 2 of 5 predefined PANSS positive subscale items (delusions, conceptual disorganization, hallucinatory behavior, grandiosity, and suspiciousness/persecution) at the initial screening assessment and at baseline for enrolled patients, and a Clinical Global Impression-Severity of Illness (CGI-S) score of 4 or higher (moderately ill) at baseline. Principal exclusion criteria were a clinically significant medical condition or abnormal laboratory or physical examination findings; diagnosis of residual-type schizophrenia, schizoaffective disorder, or coexisting psychiatric disorder coded on Axis I; current or past substance abuse; 20% or higher decrease in PANSS total score from screening to baseline; known allergy or sensitivity to haloperidol; imminent risk of self-harm or harm to others; and previous participation in an asenapine trial.
The study consisted of an up to 3-day screening period, which included a screening visit, washout period, and baseline visit, and 6 weeks of active treatment. Patients were randomly assigned to double-blind fixed-dose treatment with asenapine at 5 mg BID, asenapine at 10 mg BID (after 1 day at 5 mg BID), placebo, or haloperidol at 4 mg BID. Patients were hospitalized for the first 2 weeks of the 6-week study. Those who were viewed clinically capable could complete the remainder of the study on an outpatient basis. For patients not clinically capable of being discharged on day 14, extended hospitalization was to be approved by the sponsor. Hospitalization beyond day 21 was to be accompanied by adequate documentation in the medical record. This documentation was to be updated every 3 to 4 days.
Efficacy Assessments
The primary efficacy endpoint was the change from baseline in the total score on the PANSS, which was administered at screening, baseline, day 4, and each week of treatment. Secondary efficacy assessments included the PANSS subscale scores (positive, negative, and general psychopathology), PANSS Marder factors (positive symptoms, negative symptoms, disorganized thought, hostility/excitement, and anxiety/depression),17 CGI-S,18 and Clinical Global Impression-Improvement (CGI-I).18 We also report the percentage of PANSS responders (defined a priori as a ?30% decrease in PANSS total score from baseline to study end) and the percentage of CGI-I responders (defined as a CGI-I score of 1 [very much improved] or 2 [much improved]). In addition, results are provided for assessments using the Calgary Depression Scale for Schizophrenia (CDSS),19 Modified International Suicide Prevention Trial (InterSePT) Scale for Suicidal Thinking,20 and Readiness to Discharge Questionnaire (RDQ).21
Safety and Tolerability Assessments
At each clinic visit, investigators obtained information on adverse events (AEs) by questioning or examining the patient. Vital signs were assessed at screening, baseline, and treatment weeks 2, 3, 4, and 6; weight was recorded at screening, baseline, and treatment weeks 2, 4, and 6. A physical examination was performed at screening and week 6; laboratory tests were performed at screening, baseline, and treatment weeks 2, 4, and 6. Extrapyramidal symptom severity was assessed using the Simpson-Angus Scale (SAS),22 Barnes Akathisia Rating Scale (BARS),23 and Abnormal Involuntary Movement Scale (AIMS).24 Assessments for EPS were made at baseline and at each week of treatment. In addition, any clinically meaningful movement disorders observed by the investigator or reported by the patient were recorded. Use of antiparkinsonian medication was also recorded.
Statistical Analysis
Safety assessments were made using data from the treated population (all patients who received ?1 dose of study medication); efficacy analyses were based on data from the intent-to-treat (ITT) population (treated patients who had ?1 postbaseline PANSS assessment).
For comparisons of active treatment versus placebo on the primary and secondary efficacy measures, 2 prespecified methodologies were performed. Analysis of covariance using last observation carried forward (LOCF), pooled investigative site and treatment as factors, and baseline score as a covariate was the primary analysis. A mixed model for repeated measures (MMRM) with an undefined covariance structure was the secondary analysis. In this analysis, pooled investigative site, treatment, visit, and a visit by treatment interaction were factors and baseline score as a covariate.
Rates of PANSS response and CGI-I response (as defined earlier) were assessed using the Cochran-Mantel-Haenszel test adjusted by pooled investigative site. Data from the CDSS and the Modified InterSePT Scale for Suicidal Thinking were analyzed using analysis of covariance. Time to RDQ results were analyzed using Kaplan-Meier survival methods.
Sample size was calculated to obtain a statistical power of at least 90%. The 2-sided significance level was ? = 0.05. Hochberg adjustment was made for the primary endpoint for the multiple comparisons of the asenapine groups with the placebo group.
Although the study was not powered for direct comparison of the active treatments, the same methodologies were used in performing post hoc comparisons of asenapine at 5 and 10 mg BID versus haloperidol, with no adjustment of P values for multiple comparisons or multiple testing.
Results
Of 513 patients screened, 458 were randomly assigned to treatment with asenapine at 5 mg BID (n = 114), asenapine at 10 mg BID (n = 106), placebo (n = 123), or haloperidol at 4 mg BID (n = 115). Of these, 3 patients from the asenapine at 5 mg BID treatment group did not receive at least 1 dose of study medication and were excluded from the treated population; 455 patients took 1 or more doses of study medication and were included in the treated population for safety analysis and 448 treated patients had 1 or more postbaseline efficacy assessment and were included in the ITT population for efficacy analysis. In all, 272 patients (61% of the ITT population) completed the study.
Discontinuation rates from the ITT population were as follows: asenapine at 5 mg BID, 41 (37.6%) of the 109 patients including 14 discontinuations due to insufficient therapeutic response (lack of efficacy plus worsening symptoms reported as an AE); asenapine at 10 mg BID, 35 (33.3%) of the 105 patients including 17 due to insufficient response; haloperidol, 47 (42.0%) of the 112 patients including 10 due to insufficient response; placebo, 53 (43.4%) of the 122 patients including 31 due to insufficient response (refer to Supplemental Figure A, Supplementary Digital Content 1, Patient Disposition).
Across the 4 treatment groups in the treated population, demographic and clinical characteristics were comparable: 52% to 68% of patients were males, 59% to 64% were white, mean age range was 37 to 40 years, mean weight was 75 to 78 kg (165-172 lb), mean body mass index was 26.0 to 26.7 kg/m2. The mean age at onset of illness was 26 years (range, 6-60 yr), 91% had a diagnosis of paranoid schizophrenia, 50% exhibited current or past prominent negative symptoms, 54% had 4 or more previous episodes of acute schizophrenia requiring hospitalization, and 29% had a history of 1 or more suicide attempt. A majority of the patients in each treatment group had a history of smoking within the past 6 months (asenapine at 5 mg BID, 49.5%; asenapine at 10 mg BID, 67.9%; placebo, 57.7%; haloperidol, 58.3%) at baseline; among the smokers, 99% smoked up to 2 packs per day.
Concomitant medications were used by 61%, 71%, 78%, and 82% of patients treated with asenapine at 5 mg BID, asenapine at 10 mg BID, placebo, and haloperidol, respectively. Medications used by at least 10% of patients in any treatment group included lorazepam (asenapine at 5 mg BID, 40%; asenapine at 10 mg BID, 43%; placebo, 49%; haloperidol, 45%), zolpidem (19%; 23%; 22%; 22%), acetaminophen (14%; 22%; 24%; 13%), trihexyphenidyl (10%; 15%; 7%; 31%), benzatropine (7%; 5%; 6%; 14%), and ibuprofen (6%; 8%; 11%; 12%).
Efficacy
On both LOCF and MMRM analyses of change in PANSS total score, asenapine at 5 mg BID and haloperidol were both superior to placebo, with statistically significant differences seen from day 21 onward (Fig. 1A); asenapine at 10 mg BID showed no advantage over placebo at any time point on the LOCF analysis and advantage only at day 42 on the MMRM analysis (Fig. 1B).
FIGURE 1. Change from baseline in PANSS total score.
A, Mean ± SE, LOCF analysis; B, LS mean ± SE, MMRM analysis. ASE 5, asenapine at 5 mg BID; ASE 10, asenapine at 10 mg BID; HAL, haloperidol at 4 mg BID; PLAC, placebo.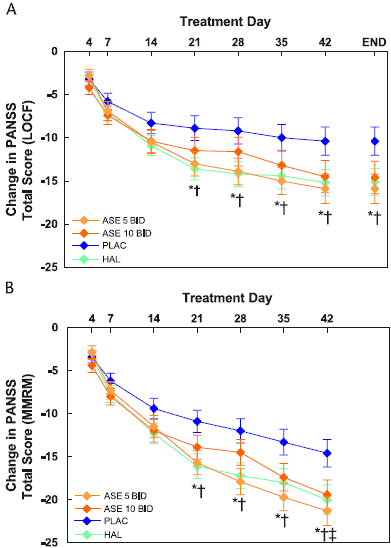
For secondary efficacy measures, on both LOCF and MMRM analyses of change in the PANSS positive subscale score, asenapine at 5 mg BID and haloperidol were superior to placebo from day 21 onward (Fig. 2A), whereas asenapine at 10 mg BID showed advantage at day 42 and study endpoint (Fig. 2B). On the LOCF analysis of change in the PANSS negative subscale, none of the treatments was superior to placebo (Fig. 2C); on the MMRM analysis, only asenapine at 5 mg BID showed advantage, at days 35 and 42 (Fig. 2D). On both LOCF and MMRM analyses of change in the PANSS general psychopathology subscale score, asenapine at 5 mg BID was superior to placebo from day 21 onward, whereas haloperidol showed advantage at day 21 only, and asenapine at 10 mg BID showed no advantage at any time point (Fig. 2E, F).
FIGURE 2. Change from baseline in PANSS subscale scores. Positive subscale: A, Mean ± SE, LOCF analysis; B, LS mean ± SE, MMRM analysis. Negative subscale: C, Mean ± SE, LOCF analysis; D, LS mean ± SE, MMRM analysis. General psychopathology subscale: E, Mean ± SE, LOCF analysis; F, LS mean ± SE, MMRM analysis.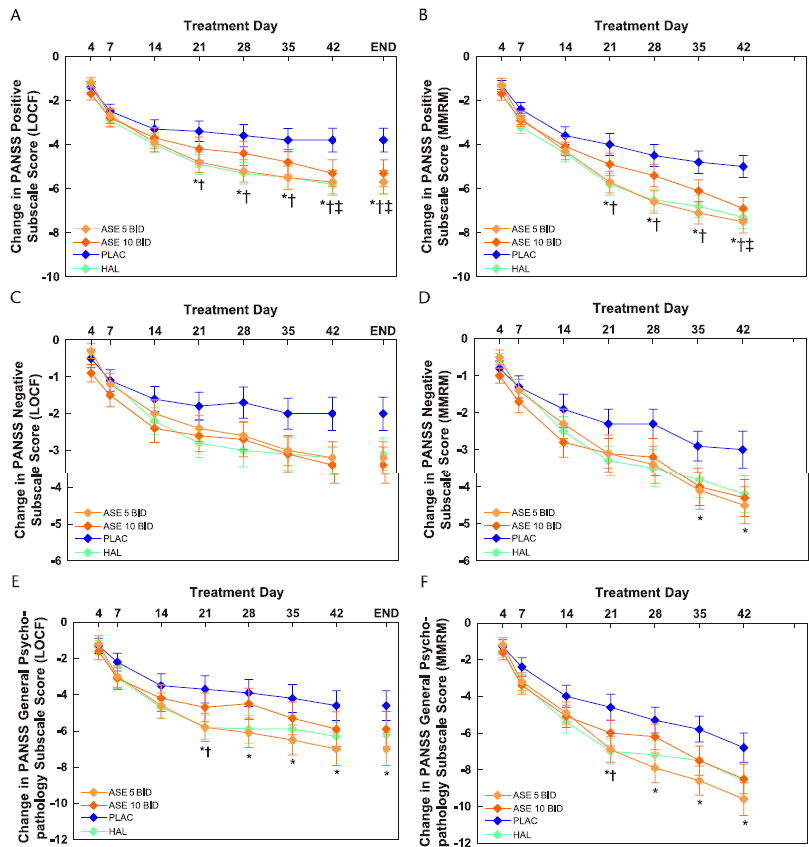
The changes noted on some of the PANSS Marder factor scores paralleled the PANSS subscale results (Table 1). On both the LOCF and MMRM analyses, all active treatment regimens were superior to placebo on the positive factor, but none showed advantage on the negative factor. On both analyses of the hostility/excitement factor, only haloperidol was superior to placebo. Asenapine at 5 mg BID was the only regimen that showed an advantage over placebo on the anxiety/depression factor (MMRM analysis only) and the disorganized thought factor (LOCF and MMRM analyses).
TABLE 1.
Changes from Baseline to Endpoint in PANSS Marder Factor Scores
| Asenapine, 5 mg BID (n = 109) | Asenapine, 10 mg BID (n = 105) | Placebo (n = 122) | Haloperidol, 4 mg BID (n = 112) | |
| Positive symptoms | ||||
| Mean ± SE baseline score | 27.1 ± 0.44 | 26.9 ± 0.47 | 27.0 ± 0.44 | 26.7 ± 0.43 |
| Mean ± SE change (LOCF) | -6.1 ± 0.59 | -5.6 ± 0.62 | -3.8 ± 0.52 | -5.7 ± 0.56 |
| LS mean ± SE change (MMRM) | -7.7 ± 0.6 | -7.3 ± 0.6 | -5.1 ± 0.6 | -7.3 ± 0.6 |
| Negative symptoms | ||||
| Mean ± SE baseline score | 21.6 ± 0.52 | 21.8 ± 0.50 | 20.9 ± 0.46 | 21.7 ± 0.54 |
| Mean ± SE change (LOCF) | -3.1 ± 0.44 | -3.5 ± 0.51 | -2.1 ± 0.51 | -2.9 ± 0.46 |
| LS mean ± SE change (MMRM) | -4.4 ± 0.5 | -4.4 ± 0.5 | -3.3 ± 0.5 | -4.0 ± 0.5 |
| Hostility/excitement | ||||
| Mean ± SE baseline score | 8.9 ± 0.31 | 9.2 ± 0.36 | 9.8 ± 0.33 | 9.2 ± 0.33 |
| Mean ± SE change (LOCF) | -1.0 ± 0.31 | -1.0 ± 0.38 | -0.8 ± 0.39 | -1.7 ± 0.35 |
| LS mean ± SE change (MMRM) | -1.8 ± 0.3 | -1.8 ± 0.3 | -1.3 ± 0.3 | -2.4 ± 0.3 |
| Anxiety/depression | ||||
| Mean ± SE baseline score | 10.3 ± 0.31 | 10.0 ± 0.32 | 10.0 ± 0.30 | 9.6 ± 0.32 |
| Mean ± SE change (LOCF) | -2.2 ± 0.33 | -1.5 ± 0.32 | -1.5 ± 0.26 | -1.6 ± 0.32 |
| LS mean ± SE change (MMRM) | -2.8 ± 0.3 | -2.3 ± 0.3 | -2.0 ± 0.3 | -2.3 ± 0.3 |
| Disorganized thought | ||||
| Mean ± SE baseline score | 21.3 ± 0.42 | 21.2 ± 0.39 | 21.2 ± 0.35 | 21.4 ± 0.50 |
| Mean ± SE change (LOCF) | -3.5 ± 0.46 | -2.9 ± 0.45 | -2.2 ± 0.40 | -3.2 ± 0.37 |
| LS mean ± SE change (MMRM) | -4.7 ± 0.4 | -4.2 ± 0.4 | -3.2 ± 0.4 | -4.2 ± 0.4 |
Values shown in italic indicate P ? 0.05 versus placebo.
The percentage of patients classified as PANSS responders at endpoint (LOCF analysis) was significantly greater with asenapine at 5 mg BID (55%, P <0.001) and 10 mg BID (49%; P <0.05) than with placebo (33%); advantage with asenapine was seen from day 21 onward in patients receiving 5 mg BID, and at day 42 and endpoint in those receiving 10 mg BID. Haloperidol showed no statistically significant advantage over placebo at day 42 (44% vs 33% with placebo) or endpoint (43% vs 33% with placebo). Similarly, the percentage of CGI-I responders at endpoint (LOCF analysis) was greater with asenapine at 5 mg BID (48%; P <0.05) than with placebo (34%); the responder rates with asenapine at 10 mg BID and haloperidol (both 44%) were not significantly higher than the rate with placebo.
On LOCF and MMRM analyses of change in the CGI-S score, asenapine at 5 mg BID and haloperidol were both significantly better than placebo from day 21 onward. At endpoint on the LOCF analysis, mean (SE) change from baseline was -0.91 ± 0.11 for asenapine at 5 mg BID and -0.88 ± 0.10 for haloperidol (both P <0.05 vs -0.59 ± 0.09 for placebo); the change with asenapine at 10 mg BID was -0.86 ± 0.11 (nonsignificant vs placebo). On the MMRM analysis, least squares (LS) mean (SE) changes from baseline were -1.2 ± 0.1 for both asenapine at 5 mg BID and haloperidol and -1.1 ± 0.1 for asenapine at 10 mg BID (all P <0.05 vs -0.8 ± 0.1 for placebo).
On LOCF analysis of change in CDSS score, significant improvement was seen with asenapine at 5 mg BID starting at day 21. At endpoint, mean (SE) change from baseline was -1.8 ± 0.31 with asenapine at 5 mg BID (P <0.05 vs -0.6 ± 0.33 with placebo); changes with asenapine at 10 mg BID and haloperidol were -1.1 ± 0.31 and -0.9 ± 0.32, respectively (both nonsignificant vs placebo).
On the Modified InterSePT Scale for Suicidal Thinking, changes from baseline to endpoint were small in all groups, and no active treatment showed significant difference from placebo. Changes on the RDQ were small in all groups, with no statistically significant differences from placebo. The Kaplan-Meier estimate indicated that 50% of patients were ready for discharge on day 15 with asenapine at 5 mg BID and haloperidol, on day 16 with asenapine at 10 mg BID, and on day 20 with placebo.
The results of post hoc comparisons between asenapine and haloperidol are summarized in Table 2. Using both LOCF and MMRM methodology, the only efficacy measure showing a statistically significant difference was the PANSS response rate at endpoint, which was 55% for asenapine at 5 mg BID versus 43% for haloperidol (LOCF analysis, P = 0.0451). On all other efficacy measures, using both methodologies, there were no significant differences between asenapine at either dosage and haloperidol.
TABLE 2.
Post Hoc Comparison of Asenapine Versus Haloperidol on Efficacy Measures (P values vs haloperidol)
| Measure | LOCF at Endpoint | MMRM at Day 42 | ||
| Asenapine, 5 mg BID | Asenapine, 10 mg BID | Asenapine, 5 mg BID | Asenapine, 10 mg BID | |
| PANSS total score (primary outcome) | 0.732 | 0.800 | 0.585 | 0.798 |
| PANSS subscales | ||||
| Positive subscale | 0.917 | 0.535 | 0.876 | 0.571 |
| Negative subscale | 0.597 | 0.531 | 0.658 | 0.828 |
| General psychopathology subscale | 0.631 | 0.685 | 0.422 | 0.843 |
| PANSS Marder factors | ||||
| Positive symptoms | 0.718 | 0.833 | 0.633 | 0.934 |
| Negative symptoms | 0.648 | 0.415 | 0.544 | 0.536 |
| Disorganized thought | 0.531 | 0.758 | 0.350 | 0.977 |
| Hostility/excitement | 0.343 | 0.191 | 0.188 | 0.235 |
| Depression/anxiety | 0.573 | 0.512 | 0.230 | 0.963 |
| Other secondary outcome measures | ||||
| CGI-Severity of Illness scale | 0.980 | 0.620 | 0.956 | 0.649 |
| CDSS* | 0.092 | 0.696 | — | — |
| PANSS response rate* | 0.0451 | 0.2971 | — | — |
| CGI-Improvement Scale, response rate* | 0.5219 | 0.9427 | — | — |
*Assessed by LOCF only.
Safety
Adverse event data, including the number of patients in each group experiencing AEs, the number who withdrew because of such events, and the most common AEs, are summarized in Table 3. Adverse events occurring in at least 5% of patients in any active treatment group and with an incidence more than twice that seen with placebo included akathisia (15% with haloperidol and 12% with asenapine at 10 mg BID vs 3% with placebo; 5% with asenapine at 5 mg BID), Parkinsonism (14% with haloperidol vs 5% with placebo; 8% and 7% with asenapine at 5 and 10 mg BID, respectively), oral hypoesthesia (11% and 9% with asenapine at 5 and 10 mg BID vs 2% with placebo; 0% with haloperidol), dystonia (10% with haloperidol vs 0% with placebo; 4% and 2% with asenapine at 5 and 10 mg BID), somnolence (9% and 8% with asenapine at 5 and 10 mg BID vs <1% with placebo; 2% with haloperidol), vomiting (7% with asenapine at 10 mg BID vs 2% with placebo; <1% with asenapine at 5 mg BID and 2% with haloperidol), and muscle rigidity (7% with haloperidol vs 0% with placebo; 3% and 4% with asenapine at 5 and 10 mg BID).
TABLE 3.
Summary of AEs
| n (%) of Patients | ||||
| Asenapine, 5 mg BID (n = 111) | Asenapine, 10 mg BID (n = 106) | Placebo (n = 123) | Haloperidol, l4 mg BID (n = 115) | |
| All AEs | 72 (65) | 80 (76) | 89 (72) | 87 (76) |
| Mild | 34 (31) | 39 (37) | 47 (38) | 33 (29) |
| Moderate | 28 (25) | 36 (34) | 36 (29) | 47 (41) |
| Severe | 10 (9) | 5 (5) | 6 (5) | 7 (6) |
| All treatment-related AEs | 49 (44) | 55 (52) | 50 (41) | 65 (57) |
| Mild | 28 (25) | 38 (36) | 31 (25) | 31 (27) |
| Moderate | 18 (16) | 15 (14) | 15 (12) | 30 (26) |
| Severe | 3 (3) | 2 (2) | 4 (3) | 4 (4) |
| Serious AEs | ||||
| All serious AEs | 7 (6) | 9 (9) | 9 (7) | 8 (7) |
| Treatment-related serious AEs | 2 (2) | 3 (3) | 6 (5) | 4 (4) |
| Discontinuations due to | ||||
| All AEs | 5 (5) | 10 (9) | 13 (11) | 12 (11) |
| Treatment-related AEs | 3 (3) | 5 (5) | 9 (7) | 8 (7) |
| Most common AEs (>5% of patients) | ||||
| Insomnia | 22 (20) | 19 (18) | 18 (15) | 16 (14) |
| Oral hypoesthesia | 12 (11) | 10 (9) | 3 (2) | 0 |
| Somnolence | 10 (9) | 8 (8) | 1 (<1) | 2 (2) |
| Agitation | 10 (9) | 7 (7) | 11 (9) | 9 (8) |
| Parkinsonism | 9 (8) | 7 (7) | 6 (5) | 16 (14) |
| Headache | 9 (8) | 6 (6) | 12 (10) | 5 (4) |
| Sedation | 7 (6) | 6 (6) | 5 (4) | 4 (4) |
| Akathisia | 6 (5) | 13 (12) | 4 (3) | 17 (15) |
| Anxiety | 5 (5) | 7 (7) | 5 (4) | 7 (6) |
| Worsening psychotic symptoms | 3 (3) | 9 (9) | 10 (8) | 8 (7) |
| Vomiting | 1 (<1) | 7 (7) | 3 (2) | 2 (2) |
| Dystonia | 4 (4) | 2 (2) | 0 | 11 (10) |
| Muscle rigidity | 3 (3) | 4 (4) | 0 | 8 (7) |
Extrapyramidal Symptoms
Extrapyramidal symptoms were reported as an AE in 15%, 18%, 10%, and 34% of patients in the asenapine at 5 and 10 mg BID, placebo, and haloperidol groups, respectively. In most cases, EPS were rated as mild or moderate. On the rating scale assessments (Table 4), patients receiving asenapine at 5 and 10 mg BID showed modest improvements on the AIMS and the SAS and virtually no change on the BARS; patients receiving haloperidol showed improvement on the AIMS and worsening on the BARS and SAS (the latter in marked contrast with improvement in the placebo group). During the study, antiparkinsonian medication was initiated in 17%, 19%, 12%, and 43% of patients in the asenapine at 5 and 10 mg BID, placebo, and haloperidol groups, respectively.
TABLE 4.
Baseline Score and Change at Endpoint on Rating Scale Assessments of EPS
| Asenapine, 5 mg BID | Asenapine, 10 mg BID | Placebo | Haloperidol, 4 mg BID | |||||
| Baseline* (n = 111) | Chang e (n = 108) | Baseline* (n = 106) | Change (n = 105) | Baseline* (n = 123) | Change (n = 120) | Baseline* (n = 115) | Change (n = 111) | |
| AIMS 7 | 0.53 ± 1.46 | -0.08 ± 1.05 | 0.41 ± 1.73 | -0.07 ± 0.62 | 0.56 ± 1.59 | 0.05 ± 1.56 | 0.74 ± 2.08 | -0.24 ± 1.69 |
| BARS Global Score | 0.19 ± 0.51 | 0.00 ± 0.51 | 0.24 ± 0.64 | 0.02 ± 0.84 | 0.28 ± 0.75 | -0.10 ± 0.73 | -0.17 ± 0.54 | 0.21 ± 0.89 |
| SAS Total Score | 1.33 ± 2.94 | -0.17 ± 3.12 | 1.32 ± 2.94 | -0.07 ± 1.98 | 1.37 ± 2.63 | -0.62 ± 2.32 | 1.41 ± 3.12 | 0.86 ± 3.13 |
*Baseline mean for patients completing the visit.
Laboratory Measures
Mean changes from baseline in levels of various laboratory measures, including lipids, fasting glucose, and liver enzymes, were small in all treatment groups and comparable to those seen with placebo. No patients in any treatment group had abnormal postbaseline total cholesterol levels (defined a priori as 50% above upper limit of normal). The percentage of patients with abnormal postbaseline fasting glucose levels (defined a priori as 50% above upper limit of normal) was 1%, 2%, 1%, and 3% in the asenapine at 5 and 10 mg BID, placebo, and haloperidol groups, respectively.
Prolactin
From baseline to last assessment, prolactin levels decreased in the placebo group and both asenapine groups but increased in the haloperidol group. The mean (SD) change in prolactin levels was -14.7 (38.7) ?g/L for asenapine at 5 mg BID, -11.6 (40.9) ?g/L for asenapine at 10 mg BID, -19.2 (43.7) ?g/L for placebo, and 2.8 (59.1) ?g/L for haloperidol. The percentage of patients with abnormal postbaseline prolactin levels (defined a priori as >4 times upper limit of normal) was 4%, 5%, 2%, and 10% in the asenapine at 5 and 10 mg BID, placebo, and haloperidol groups, respectively.
Weight
Incidence rates of clinically significant weight gain (?7% increase from baseline) were 5%, 4%, 2%, and 4% in the asenapine at 5 and 10 mg BID, placebo, and haloperidol groups, respectively; incidence rates of clinically significant weight loss (? 7% decrease from baseline) were 1%, 2%, 2%, and 0%. The mean (SD) change in weight from baseline to endpoint was 0.7 (2.5) kg [1.5 (5.5) lb] for asenapine at 5 mg BID, 0.6 (2.5) [1.3 (5.5) lb] kg for asenapine at 10 mg BID, -0.4 (5.8) kg [-0.9 (12.8) lb] for placebo, and 0.3 (2.4) kg [0.7 (5.3) lb] for haloperidol.
Discussion
In this study, using LOCF analysis, asenapine at 5 mg BID was effective in the treatment of acute schizophrenia, as indicated by significant improvements in PANSS total score, PANSS positive and general psychopathology subscale scores, PANSS Marder factor positive and disorganized thought scores, and the percentage of PANSS responders. Results with MMRM analysis with asenapine 5 mg BID were supportive of the LOCF results. Based on previously published reports of 4- to 6-week studies in patients with schizophrenia, the effects of asenapine and the active comparator haloperidol are of a magnitude that is generally comparable with those of currently marketed antipsychotics. 13,25 33 Although it is not clear why statistically significant differences were not observed with any treatment arm on the RDQ, estimates for readiness to discharge were generally shorter with active treatment (15-16 days) compared with placebo (20 days).
Reductions in PANSS total score with asenapine at 10 mg BID did not achieve statistical significance compared with placebo using LOCF analysis, even though the magnitude of the reduction with asenapine at 10 mg BID was roughly comparable with those of asenapine 5 mg BID and haloperidol and numerically greater than that of placebo. The reasons for differences in the statistical outcomes and the lack of a dose response between the 2 asenapine dosage groups are unclear. There were no major differences in demographic or clinical characteristics between these groups. Treatment-emergent AEs were more frequent with asenapine at 10 mg BID, but this finding does not constitute evidence of dose-relationship; indeed, discontinuations due to AEs and discontinuations due to insufficient therapeutic response were both comparable between the 2 dosage groups. One potential factor that might contribute to the lack of statistically significant therapeutic effect with asenapine at 10 mg BID is the high placebo response rate seen in this trial. Using LOCF analysis, the reduction in PANSS total score with placebo was 10.7 points, a value that exceeds placebo response rates reported in other studies (range, 3.3 to -7.1 points).13,25 33 When the placebo response is substantial, even a slight difference in the magnitude of response with active treatment could result in separation from placebo for 1 regimen but not the other. The fact that asenapine at 10 mg BID did separate from placebo with MMRM, an analysis technique that may more appropriately account for missing data,34 supports this possibility.
In general, our study supports the findings of an earlier trial of asenapine in acute schizophrenia reported by Potkin et al.13 In that study, 182 patients were randomly assigned to 6 weeks of BID treatment with asenapine at 5 mg, placebo, or risperidone at 3 mg. At endpoint on LOCF analysis, asenapine at 5 mg was significantly better than placebo in improving the PANSS total score and general psychopathology subscale score. Adverse events, including EPS, in the asenapine group were comparable to those in the placebo group.
In our study, improvements were also seen with asenapine at 5 mg BID on both the CDSS and the PANSS anxiety/depression Marder factor. This is of clinical relevance because depressive symptoms have been reported in about 50% of newly diagnosed patients with schizophrenia and in about 30% of those who have relapsed.35 In addition to complicating the treatment of schizophrenia, depressive symptoms are one of several significant risk factors for suicide among patients with schizophrenia.36 Some atypical antipsychotics appear to have a direct effect on depressive symptoms in schizophrenia.37 However, baseline levels of depression in our study and the study of Potkin et al13 were low, making it more difficult to discern any change in depression. Further studies will be needed to evaluate asenapine for depressive symptoms in patients with schizophrenia.
Our post hoc analysis of efficacy with asenapine versus haloperidol revealed no significant differences, except for a higher PANSS response rate at endpoint with asenapine at 5 mg BID than with haloperidol. Although these findings should be interpreted in light of the fact that the study was not powered for direct comparison of the active treatments, they are consistent with studies showing overlaps between conventional and atypical antipsychotics in terms of efficacy,38 and small-to-medium effect-size advantages with some atypicals but not others.39
Both asenapine doses were generally safe and well tolerated, with most AEs seen with asenapine rated as mild or moderate. The incidence of serious AEs and that of AEs leading to discontinuation with asenapine were comparable to that of placebo and haloperidol, with rates being slightly lower with asenapine at 5 mg BID than with asenapine at 10 mg BID. In general, the AE profiles of asenapine at 5 mg BID and 10 mg BID were similar except for a higher incidence of akathisia in the asenapine at 10 mg BID group. Neither asenapine at 5 mg BID nor 10 mg BID had any notable effect on metabolic variables.
A lower risk of EPS is a distinguishing feature of atypical antipsychotics compared with conventional agents, yet even with the newer agents, patients still discontinue treatment because of EPS. In the Clinical Antipsychotic Trials of Intervention Effectiveness, EPS were the cause of discontinuation for 11% of patients who discontinued for any reason and for 27% of those who discontinued because of an AE.3 The incidence of EPS reported as an AE and as measured on the SAS with both asenapine doses was considerably lower than that observed with haloperidol in our study; a finding that is not unexpected given that atypical antipsychotics typically have lower risk for EPS than conventional antipsychotics. Also, the need for antiparkinsonian medication during the study was higher in the haloperidol group than in either asenapine group. Although the dosage of haloperidol used in our study is not considered excessive in terms of EPS liability40 and is reasonable considering the mean severity of symptoms observed in this population at baseline, it is possible that a lower dosage might have been associated with a lower incidence of EPS. These findings are similar to those reported in comparisons of olanzapine and aripiprazole with haloperidol, in which the incidence of EPS and need for antiparkinsonian medication was higher with haloperidol than with the atypical antipsychotics.3
Change in weight with asenapine was minimal, with 5% or fewer patients in either asenapine group experiencing clinically significant weight gain or weight loss. These results are similar to those seen in the Potkin et al13 study, in which the incidence of clinically significant weight gain was 4%.
Hyperprolactinemia did not develop in our patients treated with asenapine, with small decreases in mean prolactin levels seen in both asenapine groups. Potkin et al13 also reported a minimal effect of asenapine on prolactin, with postbaseline levels 2 or more times the upper limit of normal reported in 9% of patients treated with asenapine compared with 79% of those given risperidone. The prolactin profile of asenapine appears to be similar to that of clozapine, olanzapine, quetiapine, and ziprasidone, which have not been associated with considerable or sustained increases in prolactin levels, and in contrast to that of risperidone, which has been associated with hyperprolactinemia at higher dosages (>4 mg/day).41
Several questions may be raised concerning the findings of this study, starting with the ethics of using placebo in patients with acute schizophrenia. Placebo remains a standard comparator to establish efficacy. Further, the fact that response rates with placebo can be surprisingly high in clinical trials assessing psychiatric illness,42,49 including schizophrenia,50,51 underscores the need to demonstrate that active treatment can separate from placebo.
A question could also be raised in relation to how these data can be generalized to a real world population because patients with current or past substance abuse and those with a diagnosis of residual schizophrenia were excluded from this study. Nevertheless, this is common practice in short-term clinical trials.
The completion rates (62%, 67%, 57%, and 58% with asenapine at 5 mg BID, asenapine at 10 mg BID, placebo, and haloperidol, respectively) were higher than those reported in the study by Potkin et al, in which fewer than half of the patients across all treatment groups completed the study. Like the earlier study, we used fixed doses instead of flexible dose adjustment based on clinical effects, which may contribute to discontinuation. High rates of discontinuation are frequently seen in schizophrenia trials, most often because of poor therapeutic response.5
In conclusion, our results show that 6 weeks of fixed-dose treatment with asenapine at 5 mg BID is effective in the treatment of acute schizophrenia, with secondary statistical analyses indicating potential efficacy at day 42 for 10 mg BID as well. Asenapine also appeared to be well tolerated in these patients. The magnitude of clinical effect with asenapine and haloperidol appeared similar, but greater contrasts were seen in AEs, especially EPS. Further studies are in progress to evaluate the long-term safety and maintenance of effect of asenapine.
Acknowledgments
Editorial services to the authors were provided by Steven Tiger, an employee of Complete Healthcare Communications, Inc, and funded by Schering-Plough Corporation, now Merck & Co, Inc, (Whitehouse Station, NJ, USA).
The authors thank the following investigators who participated in this trial: Yuri Alexandrovsky, Moscow, Russia; Daniel Anderson, Torrance, California, USA; Petru Boisteanu, Iasi, Romania; Gary Booker, Shreveport, Louisiana, USA; Guy Brannon, Shreveport, Louisiana, USA; Saroj Brar, Cleveland, Ohio, USA; Mikhail Burdukovsky, St. Petersburg, Russia; Victoria Burtea, Brasov, Romania; Norman Costigan, Red Deer, Alberta, Canada; Irina Dan, Bucuresti, Romania; Himasiri De Silva, Santa Ana, California, USA; Lakshman Dutt, Ahmedabad, Gujarat, India; Carlos Figueroa, Pasadena, California, USA; David Flaherty, North Miami, Florida, USA; Donald Garcia, Austin, Texas, USA; Ramanath Gopalan, Arlington, Virginia, USA; Elena Grigorieva, Yaroslavl, Russia; Monica Ienciu, Timisoara, Romania; Richard Jaffe, Philadelphia, Pennsylvania, USA; Lala Kasimova, Nizhniy Novgorod, Russia; Alexander Kociubynski, St. Petersburg, Russia; Joseph Kwentus, Flowood, Mississippi, USA; Herbert Meltzer, Nashville, Tennessee, USA; Alexander Mouzitchenko, Moscow, Russia; Rajesh Nagpal, New Delhi, India; Nikolay Neznanov, St. Petersburg, Russia; Gheorghe Oros, Oradea, Romania; Sanjay Phadke, Pune, Mahara, India; Mikhail Popov, St. Petersburg, Russia; Dan Prelipceanu, Bucuresti, Romania; N. N. Raju, Visakhapatnam, Andh Prad, India; Evgenia Rebrova, St. Petersburg, Russia; Alexander Reznik, Hotkovo, Russia; Ramanathan Sathianathan, Chennai, Tamilnadu, India; Scott Segal, North Miami, Florida, USA; P.S. V N. Sharma, Manipal, Karna, India; Mikhail Sheifer, Samara, Russia; Anantha Shekhar, Indianapolis, Indiana, USA; Franco Sicuro, St. Louis, Missouri, USA; Bart Sloan, Darien, Connecticut, USA; T. P. Sudhakar, Tirupati, Tamilnadu, India; Daniel Vasile, Bucuresti, Romania; Kausar Yakhin, Kazan, Russia; Daniel Zimbroff, Upland, California, USA.
Author disclosure information
Dr Kane has served as a consultant or participated on advisory boards for Bristol-Myers Squibb, Otsuka Pharmaceuticals, Eli Lilly and Co, Janssen, Johnson & Johnson PRD, MDS Pharma Services, Pfizer Inc, Solvay Pharmaceuticals, Inc, Wyeth Pharmaceutical, Lundbeck, Vanda Pharmaceutical, Astra-Zeneca, Cephalon, Dainippon Sumitomo, GlaxoSmithKline, Intracellular Therapeutics, PGxHealth, Proteus, Takeda, and Schering-Plough. Drs Cohen, Zhao, and Panagides are full-time employees of Merck. Dr Larry Alphs was employed at Pfizer Inc at the time the study was completed.
REFERENCES
1. Lieberman JA. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia: efficacy, safety and cost outcomes of CATIE and other trials. J Clin Psychiatry. 2007;68(2):e04.
2. Murphy BP, Chung YC, Park TW, et al. Pharmacological treatment of primary negative symptoms in schizophrenia: a systematic review. Schizophr Res. 2006;88(1-3):5-25.
3. Weiden PJ. EPS profiles: the atypical antipsychotics are not all the same. J Psychiatr Pract. 2007;13(1):13-24.
4. McIntosh AM, Conlon L, Lawrie SM, et al. Compliance therapy for schizophrenia. Cochrane Database Syst Rev. 2006;3:CD003442.
5. Liu-Seifert H, Adams DH, Kinon BJ. Discontinuation of treatment of schizophrenic patients is driven by poor symptom response: a pooled post-hoc analysis of four atypical antipsychotic drugs. BMC Med. 2005;3:21.
6. Droulout T, Liraud F, Verdoux H. Relationships between insight and medication adherence in subjects with psychosis. Encephale. 2003;29(5):430-437.
7. Haro JM, Suarez D, Novick D, et al. Three-year antipsychotic effectiveness in the outpatient care of schizophrenia: observational versus randomized studies results. Eur Neuropsychopharmacol. 2007;17(4):235-244.
8. Perkins DO, Gu H, Weiden PJ, et al. Predictors of treatment discontinuation and medication nonadherence in patients recovering from a first episode of schizophrenia, schizophreniform disorder, or schizoaffective disorder: a randomized, double-blind, flexible-dose, multicenter study. J Clin Psychiatry. 2008;69(1):106-113.
9. Shahid M, Walker G, Zorn S, et al. Asenapine: a novel psychopharmacologic agent with a unique human receptor signature. J Psychopharmacol. 2009;23(1):65-73.
10. Saphris®. Schering-Plough, Kenilworth, NJ. 2009.
11. Peeters PAM, Bockbrader H, Spaans E, et al. Asenapine Pharmacokinetics: Influence of Hepatic and Renal Impairment. Presented at: American Psychiatric Association; May 3-8, 2008; Washington, DC.
12. Dogterom P, Hulskotte EGJ, Gerrits MGF, et al. Asenapine Pharmacokinetics: Influence of Cytochrome P450 Modulators and UDP-Glucuronyltransferase Inhibition. Presented at: 21st Congress European College of Neuropsychopharmacology; February 3-7, 2008; Barcelona, Spain.
13. Potkin SG, Cohen M, Panagides J. Efficacy and tolerability of asenapine in acute schizophrenia: a placebo- and risperidone-controlled trial. JClin Psychiatry. 2007;68(10):1492-1500.
14. Luft B, Taylor D. A review of atypical antipsychotic drugs versus conventional medication in schizophrenia. Expert Opin Pharmacother. 2006;7(13):1739-1748.
15. Jones PB, Barnes TR, Davies L, et al. Randomized controlled trial of the effect on Quality of Life of second- vs first-generation antipsychotic drugs in schizophrenia: Cost Utility of the Latest Antipsychotic Drugs in Schizophrenia Study (CUtLASS 1). Arch Gen Psychiatry. 2006;63(10):1079-1087.
16. Kay SR, Fiszbein A, Opler LA. The Positive and Negative Syndrome Scale (PANSS) for schizophrenia. Schizophr Bull. 1987;13(2):261-276.
17. Marder SR, Davis JM, Chouinard G. The effects of risperidone on the five dimensions of schizophrenia derived by factor analysis: combined results of the North American trials. J Clin Psychiatry. 1997;58(12):538-546.
18. Guy W. Clinical global impressions. In: Guy W, ed. ECDEU Assessment Manual for Psychopharmacology. Washington, DC: U.S. Department of Health, Education, and Welfare; 1976:217-222.
19. Addington D, Addington J, Schissel B. A depression rating scale for schizophrenics. Schizophr Res. 1990;3(4):247-251.
20. Lindenmayer JP, Czobor P, Alphs L, et al. The InterSePT scale for suicidal thinking reliability and validity. Schizophr Res. 2003;63(1-2):161-170.
21. Potkin SG, Gharabawi GM, Greenspan AJ, et al. Psychometric evaluation of the Readiness for Discharge Questionnaire. Schizophr Res. 2005;80(2-3):203-212.
22. Simpson GM, Angus JW. A rating scale for extrapyramidal side effects. Acta Psychiatr Scand Suppl. 1970;212:11-19.
23. Barnes TR. A rating scale for drug-induced akathisia. Br J Psychiatry. 1989;154:672-676.
24. Guy W. Abnormal involuntary movement scale. In: Guy W, ed. ECDEU Assessment Manual for Psychopharmacology. Washington, DC: U.S. Department of Health, Education, and Welfare; 1976: 534-537.
25. Arvanitis LA, Miller BG. Multiple fixed doses of ''Seroquel'' (quetiapine) in patients with acute exacerbation of schizophrenia: a comparison with haloperidol and placebo. The Seroquel Trial 13 Study Group. Biol Psychiatry. 1997;42(4):233-246.
26. Beasley CM Jr, Hamilton SH, Crawford AM, et al. Olanzapine versus haloperidol: acute phase results of the international double-blind olanzapine trial. Eur Neuropsychopharmacol. 1997;7(2):125-137.
27. Beasley CM Jr, Sanger T, Satterlee W, et al. Olanzapine versus placebo: results of a double-blind, fixed-dose olanzapine trial. Psychopharmacology (Berl). 1996;124(1-2):159-167.
28. Beasley CM Jr, Tollefson G, Tran P, et al. Olanzapine versus placebo and haloperidol: acute phase results of the North American double-blind olanzapine trial. Neuropsychopharmacology. 1996; 14(2):111-123.
29. Cutler AJ, Kalali AH, Weiden PJ, et al. Four-week, double-blind, placebo- and ziprasidone-controlled trial of iloperidone in patients with acute exacerbations of schizophrenia. J Clin Psychopharmacol. 2008;28(2 suppl 1):S20-S28.
30. Daniel DG, Zimbroff DL, Potkin SG, et al. Ziprasidone 80 mg/day and 160 mg/day in the acute exacerbation of schizophrenia and schizoaffective disorder: a 6-week placebo-controlled trial. Ziprasidone Study Group. Neuropsychopharmacology. 1999;20(5): 491-505.
31. Davidson M, Emsley R, Kramer M, et al. Efficacy, safety and early response of paliperidone extended-release tablets (paliperidone ER): results of a 6-week, randomized, placebo-controlled study. Schizophr Res. 2007;93(1-3):117-130.
32. Kane J, Canas F, Kramer M, et al. Treatment of schizophrenia with paliperidone extended-release tablets: a 6-week placebo-controlled trial. SchizophrRes. 2007;90(1-3):147-161.
33. Marder SR, Meibach RC. Risperidone in the treatment of schizophrenia. Am J Psychiatry. 1994;151(6):825-835.
34. Barnes SA, Mallinckrodt CH, Lindborg SR, et al. The impact of missing data and how it is handled on the rate of false-positive results in drug development. Pharm Stat. 2008;7(3):215-225.
35. Whitehead C, Moss S, Cardno A, et al. Antidepressants for people with both schizophrenia and depression. Cochrane Database Syst Rev. 2002;(2):CD002305.
36. Perenyi A, Forlano R. Suicide in schizophrenia. Neuropsychopharmacol Hung. 2005;7(3):107-117.
37. Moller HJ. Antidepressive effects of traditional and second generation antipsychotics: a review of the clinical data. Eur Arch Psychiatry Clin Neurosci. 2005;255(2):83-93.
38. Volavka J, Citrome L. Oral antipsychotics for the treatment of schizophrenia: heterogeneity in efficacy and tolerability should drive decision-making. Expert Opin Pharmacother. 2009;10(12):1917-1928.
39. Leucht S, Corves C, Arbter D, et al. Second-generation versus first-generation antipsychotic drugs for schizophrenia: a meta-analysis. Lancet. 2009;373(9657):31-41.
40. Waraich PS, Adams CE, Roque M, et al. Haloperidol dose for the acute phase of schizophrenia. Cochrane Database Syst Rev. 2002;(3):CD001951.
41. Hamner M. The effects of atypical antipsychotics on serum prolactin levels. Ann Clin Psychiatry. 2002;14(3):163-173.
42. Laporte JR, Figueras A. Placebo effects in psychiatry. Lancet. 1994;344(8931):1206-1209.
43. Stein DJ, Baldwin DS, Dolberg OT, et al. Which factors predict placebo response in anxiety disorders and major depression? An analysis of placebo-controlled studies of escitalopram. J Clin Psychiatry. 2006;67(11):1741-1746.
44. Stolk P, Ten Berg MJ, Hemels ME, et al. Meta-analysis of placebo rates in major depressive disorder trials. Ann Pharmacother. 2003;37(12):1891Y1899.
45. Walsh BT, Seidman SN, Sysko R, et al. Placebo response in studies of major depression: variable, substantial, and growing. JAMA. 2002;287(14):1840Y1847.
46. Flament MF, Bisserbe JC. Pharmacologic treatment of obsessive-compulsive disorder: comparative studies. J Clin Psychiatry. 1997;58(suppl 12):18Y22.
47. Sysko R, Walsh BT. A systematic review of placebo response in studies of bipolar mania. J Clin Psychiatry. 2007;68(8): 1213Y1217.
48. Keck PE Jr, Welge JA, Strakowski SM, et al. Placebo effect in randomized, controlled maintenance studies of patients with bipolar disorder. Biol Psychiatry. 2000;47(8):756Y761.
49. Keck PE Jr, Welge JA, McElroy SL, et al. Placebo effect in randomized, controlled studies of acute bipolar mania and depression. Biol Psychiatry. 2000;47(8):748Y755.
50. Welge JA, Keck PE Jr. Moderators of placebo response to antipsychotic treatment in patients with schizophrenia: a meta-regression. Psychopharmacology (Berl). 2003;166(1):1-10.
51. Sanger TM, Tohen M, Vieta E, et al. Olanzapine in the acute treatment of bipolar I disorder with a history of rapid cycling. J Affect Disord. 2003;73(1-2):155-161.



