Дисплазия соединительной ткани, клеточная биология и молекулярные механизмы воздействия магния
СтатьиИ.Ю. Торшин, О.А. Громова
МГУ имени М.В. Ломоносова, Москва
Российский сателлитный центр Института микроэлементов ЮНЕСКО, Москва
Феноменология дисплазий соединительной ткани
Составляя около 50% массы тела, соединительная ткань (СТ) является одним из четырех основных типов ткани в традиционных классификациях (в дополнение к эпителиальной, мышечной и нервной ткани). Основная функция СТ – это структурная поддержка других тканей. Хрящ и кость являются основными разновидностями соединительной ткани, другие типы включают ареолярную соединительную ткань, скрепляющую органы, и плотную соединительную ткани, формирующую связки и сухожилия.
Недифференцированная дисплазия соединительной ткани (нДСТ) представляет собой разнородную группу заболеваний которые, в свою очередь, могут приводить к различным хроническим болезням. нДСТ часто соответствует абнормальным структурным и функциональным изменениям CT. Это приводит к нарушениям морфологии и функций органов [1–3]. Клинико-морфологические проявления нДСТ необычайно разнообразны. Они могут включать скелетные изменения, связанные с нарушением строения хряща, непропорционально длинные конечности, арахнодактилию, деформации грудной клетки, сколиозы позвоночника, плоскостопие, патологию развития зубов, прикуса, кисты, патологию суставов (склонность к вывихам), гиперэластичность, истончение, склонность к травматизации кожи, расширение вен и внешние признаки ускоренного старения – раннее формирование морщин, деформация овала лица, в том числе т.н. гравитационный птоз (обвисание мягких тканей лица). Кроме того, ДСТ предрасполагает к бронхолегочным и реноваскулярным патологиям, способствует потере мышечной массы (в том числе сердечной и глазодвигательной мускулатуры, что приводит к кардиоваскулярным и офтальмологическим патологиям [4]) и нарушениям функции желудочно-кишечных органов [5–7]. Поражения сердечно-сосудистой системы весьма разнообразны: пролапс митрального клапана (наиболее распространенная из всех сердечных аномалий при ДСТ [1] обнаруживается, как правило, при ЭхоКГ исследовании), венозная недостаточность, варикозная болезнь [8,9], а также патологии гемостаза [10].
Диагностика ДСТ базируется на этих симптомах и дополнительных данных (например, антропометрия, внешнее дыхание, уменьшенный размер сердца, сниженное артериальное давление, плетизмография, специфические характеристики ЭКГ и ультразвукового флебосканирования) [8,11]. Согласно анализу этих фенотипических маркеров нДСТ ее распространение может быть сравнительно большим среди общего населения (например, 8,5% в выборке из 400 человек [12]). Хотя часто говорится, что этиология ДСТ имеет генетический компонент, исчерпывающего анализа относительных ролей факторов окружающей среды (питание, экологическая обстановка, гигиена движения, психоэмоциональный фон) и генетических факторов не проводился. Как правило, заболевания, связанные с генетическими дефектами в COL11A1, COL2A1 и других коллагенах (например, синдром Стиклера, номер 108300 по базе данных OMIM, www.ncbi.nlm.nih.gov/omim/), характеризуются значительно более серьезной симптоматикой и, кроме того, встречаются весьма редко (не более 1 случая на 10 тыс. человек). Можно сказать, что молекулярные механизмы вовлеченные в этиологию дисплазий соединительной ткани, практически не исследованы.
Ранняя диагностика нДСТ, особенно у детей, позволяет осуществлять соответствующую реабилитационную терапию и предотвращать прогрессирование заболевания. Одним из наиболее ярких терапевтических результатов является эффективное лечение детей с ДСТ (главным образом с пролапсом митрального клапана) при помощи магний–содержащего препарата магнезиуморотата (Магнерот), а также других препаратов [13]. Анализ соотношения между концентрацией Mg2+ и ДСТ на уровне индивидуальных молекулярных генов может привести к полезным для практикующего врача выводам применительно к лечению ДСТ. Данный анализ также важен для фундаментального понимания патофизиологии и этиологии ДСТ.
Термин «дисплазия» обозначает абнормальный рост/развитие ткани или органа. Диагноз ДСТ ставится на основе тщательного анализа симптомов и результатов клинических исследований. Тем не менее диагноз ДСТ на практике редко сопровождается какими–либо конкретными гистологическими подтверждениями. Соответственно дисплазия, обнаруженная на клиническом уровне, может соответствовать многочисленным изменениям в структуре ткани.
В случае соединительной ткани дисплазия (т.е. «абнормальный рост») может происходить вследствие: 1) абнормального синтеза или сборки коллагена; 2) синтеза абнормального коллагена; 3) чрезмерной деградации коллагена; 4) нарушений структуры коллагеновых волокон, вследствие недостаточной поперечной сшивки; 5) аналогичных аномалий, связанных с эластиновыми волокнами; 6) разрушения ткани посредством аутоимунных реакций; 7) многих других, не изученных на сегодняшний момент механизмов.
Далее мы систематически рассмотрим молекулярно–клеточную биологию соединительной ткани, молекулярные механизмы гомеостаза магния, взаимосвязь концентраций магния и состояния метаболизма соединительной ткани.
Молекулярно–клеточная биология соединительной ткани
Термин «соединительная ткань» принадлежит в основном к области физиологии и биомедицины. Для того чтобы приблизиться к молекулярным механизмам, которые могут быть вовлечены в ДСТ, мы должны оперировать терминами молекулярной биологии клетки, такими как «внеклеточная матрица», «межклеточная адгезия», «адгезия матрица–клетка», «протеогликаны» и др.
Сложность любой ткани человеческого тела при рассмотрении на молекулярном уровне просто ошеломляет, и в этом отношении соединительная ткань вовсе не исключение. Часто говорится, что основные компоненты соединительной ткани – это коллагеновые волокна, гибкие волокна, клетки и ремоделирующие ферменты. Но такого рода описания не отражают фактическую сложность ткани на молекулярном уровне и реальное число вовлеченных генов. Поэтому данный раздел представляет собой сжатое резюме результатов исследований макромолекул, лежащих в основе ультраструктуры соединительной ткани с целью формулировки молекулярных интерпретаций ДСТ.
Принципиальное отличие соединительной ткани от любого другого типа ткани – это избыток внеклеточной матрицы при сравнительно небольшом числе клеток, составляющих ткань. В молекулярной биологии внеклеточная матрица (ВКМ) определена, как сложная сеть, сформированная многочисленными структурными макромолекулами (такими как протеогликаны, коллагены, и эластин). Взаимодействуя друг с другом и с клетками, эти структурные макромолекулы поддерживают структурную целостность тканей [14]. Именно матрица обеспечивает организованную среду, в пределах которой мигрирующие клетки могут перемещаться и взаимодействовать друг с другом. Все макромолекулы, составляющие ВКМ, производятся клетками в матрице. В большинстве соединительных тканей матричные макромолекулы синтезируются фибробластами, а в специализированных типах соединительной ткани, таких как, например, хрящ и кость – хондробластами и остеобластами. ВКМ состоит из трех принципиальных компонентов: гелеобразной среды, коллагеновых волокон и эластиновых волокон.
Гелеобразная среда. Наиважнейший компонент внеклеточной матрицы – это гелеобразная среда, формируемая протеогликанами: чрезвычайно растянутыми полипептидными цепями с многочисленными полисахаридными цепями глюкозаминогликанов, присоединенных по средством ковалетных связей (рис. 1).
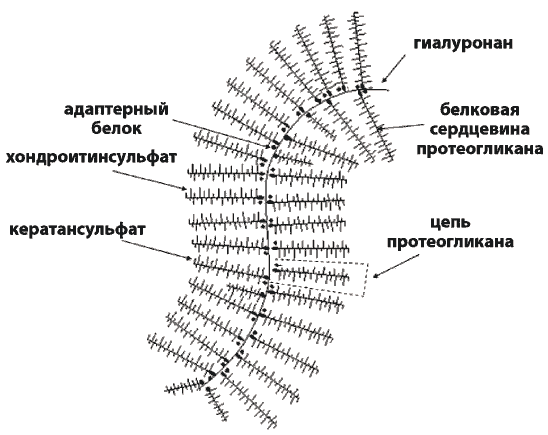
Рис. 1. Основные компоненты протеогликановых комплексов
Многочисленные цепи протеогликанов прикрепляются к особому виду глюкозаминогликана – полимеру гиалуроновой кислоты, называемому гиалуронаном. Нити гиалуронана скрепляют структуру геля в единое целое, и этот полисахаридный «гель» может противостоять сжатию и растяжению ВКМ и в то же время обеспечивать быструю диффузию питательных веществ, строительных материалов и гормонов между кровью и клетками соединительной ткани.
Механически структура геля усилена посредством волокон трех основных типов: 1) коллагенoвых волокон (состоящих главным образом из коллагена I), которые формируют скелет соединительной ткани; 2) гибких волокон (состоящих в основном из эластина и фибриллинов), которые придают соединительной ткани эластичность; 3) сетчатых (или ретикулярных) волокон (коллаген III), которые образуют перекрестные связи между всеми другими волокнами и держат вместе все остальные компоненты ткани.
Гелеобразная субстанция ВКМ сформирована протеогликанами и многодоменными гликобелками. Протеогликаны прикрепляются к нитям гиалуронана, каждая из которых содержит более 25 тыс. мономеров гиалуроновой кислоты, каждая нить может иметь длину несколько десятков микрон. Гиалуронан синтезируется посредством гиалуронансинтетаз (гены HAS1, HAS2 и HAS3) и деградируется посредством гиалуронидаз (гены HYAL2, HYAL3, HYAL4 и HYALP).
Протеогликаны содержат длинные цепочки полисахаридов, которые при физиологических условиях отрицательно заряжены вследствие ковалентно присоединенных сульфатов и уронатов. Для каждого типа протеогликана есть многочисленные белки, которые специфически связываются с этим протеогликаном, а также не менее многочисленные синтетазы, вовлеченные в синтез цепей глюкозаминогликанов и их присоединение в белковой сердцевине. Протеогликаны и соответствующие гены классифицируются согласно их глюкозаминогликанным цепям, и основные типы включают хондроитинсульфат протеогликан (гены CSPG1, CSPG2, CSPG3, CSPG4, CSPG5, CSPG6) и гепарансульфат протеогликан (перлекан, ген HSPG2). Эти протеогликаны вовлечены в основном в образование основной структуры геля. Мутации в хондроитинсульфат протеогликанах могут приводить к скелетной дисплазии [15]. Гепаран сульфат протеогликан участвует в клеточной адгезии, обладает ангиогенными свойствами и генетические дефекты в гене перлекана приводят к хронической миотонии и скелетной дисплазии [16]. Декорин (ген DCN) и люмикан (ген LUM) взаимодействуют с коллагеновыми волокнами и ограничивают диаметр волокон (см. ниже). Кератансульфатный протеогликан люмикан встречается в роговице и интерстициальных ВКМ сердца, аорты, скелетной мускулатуры, кожи и межпозвоночных дисков. Трансгенные мыши с дефицитом люмикана демонстрировали уменьшение в жесткости сухожилий, повышенную склонность к вывихам суставов и остеоартрит [17].
Ферменты, участвующие в биохимических модификациях и присоединении глюкозаминогликанов, могут значительно влиять на структуру внеклеточной матрицы. Например, дефицит ксилозил β–1,4–галактозилтрансферазы 7 (ген B4GALT7) связан с одной из форм синдрома Элерса–Данло (склонность к вывихам, хрупкая или гиперэластичная кожа, хрупкие кровяные сосуды и т.д., номер по OMIM 130000) [18].
Многодоменные гликобелки включают фибронектин, ламинины и тенасцины. Данные белки состоят из десятков однородных структурных модулей (доменов), ковалентно связанных друг с другом с образованием цепей. Фибронектин (ген FN1) участвует в адгезии клеток к ВКМ, а также в процессах миграции клеток, заживления ран, свертывания крови и иммунного ответа. Ламинины (гены LAMA1, LAMA2, LAMA3, LAMA4; LAMB1, LAMB2; LAMC1, LAMС2) важны для дифференцирования, адгезии и миграции клеток; генетические дефекты в большинстве ламининов приводят к серьезным формам мускульной дистрофии и буллезного эпидермолиза. Тенасцины (TNXB, TNC), вероятно, играют структурную роль, и дефекты в тенасцинах могут проводить к синдрому Элерса–Данло [19].
Коллагеновые волокна придают соединительной ткани прочность и долговечность. Каждое коллагеновое волокно имеет несколько микрометров в диаметре и состоит из тысяч индивидуальных полипептидных цепей коллагенов, плотно упакованных вместе. Коллагены (рис. 2) – одни из наиболее обильных белков во внеклеточной матрице и в соединительной ткани. В геноме человека существует около 50 генов, кодирующих различные коллагены, и продукты этих генов образуют более чем 20 типов коллагеновых волокон, найденных в самых различных тканях.
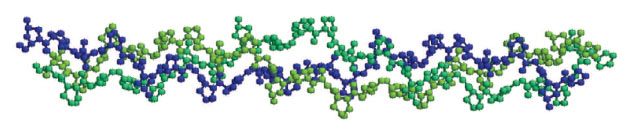
Рис. 2. Тройная спираль коллагена, основного компонента коллагеновых волокон (PDB код 1cag)
Коллагены различаются по их положению в ткани и по своему функциональному значению. Четыре основных типа коллагенов (I–IV) включают следующие гены: коллаген I (гены COL1A1, COL1A2) – основной компонент кости, который также присутствует в шрамах, сухожилиях и хрящах; коллаген II (ген COL2A1) – основной компонент хряща; коллаген III (ген COL3A1) формирует ретикулярные волокна, которые держат вместе внеклеточную матрицу; коллаген IV (гены COL4A1, COL4A2, COL4A3, COL4A4, COL4A5, COL4A6) формирует базальную ламину, на которой держится эпителий. Унаследованные и редко встречающиеся мутации в этих и других коллагенах в большинстве случаев приводят к болезни Элерса–Данло и булезному эпидермолизу.
Коллагеные болезни могут возникать не только вследствие генетических дефектов в коллагенах, но также вследствие генетических дефектов, влияющих на биосинтез, посттрансляционные модификации, секрецию, самосборку и ремоделирование коллагенов. Все эти процессы, рассмотренные ниже, поддерживаются многими дополнительными белками и генами.
За синтезом проколлагенов на рибосоме следует гидроксилирование специфических пролинов и лизинов. Эти посттрансляционные модификации зависят от аскорбиновой кислоты и необходимы для правильной сборки полипептидных цепей коллагена в коллагеновые фибрилы. Гидроксилирование пролинов катализируется пролил–3–гидроксилазой 1 (ген LEPRE1/P3H1) и мутации в этом гене вызывают рецессивное метаболическое расстройство костей [20]. Коллагены также содержат гидроксилированные лизины, к которым ковалентно прикрепляются полисахариды, стабилизирующие их структуру. Гидроксилирование поддерживается лизингидроксилазами (гены PLOD1, PLOD2, PLOD3), и мутации в этих генах приводят к дисплазии Элерса–Данло [21] и метаболическому расстройству костей [22]. Металлопротеиназа ADAMTS2 вырезает N–пропептид у проколлагенов типов I, II, V, и мутации в этом гене также вызывают болезнь Элерса–Данло [23].
Полипептидные цепи коллагена самособираются в коллагеновые фибрилы, которые впоследствии ассоциируются в коллагеновые волокна. Лизилоксидаза (ген LOX), а также лизилоксидазоподобные ферменты (ге ны LOXL1, LOXL2, LOXL3 и LOXL4) осуществляют поперечную сшивку полипептидных цепей коллагена, таким образом усиливая механическую прочность фибрил. Дефицит активности LOX обнаруживался у пациентов с синдромом Элерса–Данло [24]. Коллагеновые волокна присоединятся к клеточным мембранам через белки–адаптеры, такие как фибронектин (ген FN1) и многочисленные интегрины (более 20 генов). Протеогликанлюмикан (ген LUM) и белок фибромодулин (FMOD) влияют на самосборку цепей коллагена, ограничивая размер фибрил. Эксперименты на животных указывают, что делеции этих генов приводят к симптомам, напоминающим болезнь Элерса–Данло и другие заболевания соединительной ткани человека [17].
Ремоделирование (то есть деградация или протеолиз) коллагеновых волокон ВКМ производится посредством матриксных металлопротеиназ (ММП). Активность различных ММП имеет чрезвычайно широкий спектр биологических последствий, поскольку они де градируют большинство компонентов внеклеточной матрицы: интерстициальные коллагены и коллагены базальной мембраны, протеогликаны, декорин, фибромодулин, фибронектин и т.д. [25,26]. В геноме человека присутствуют не менее 200 ММП–подобных генов, включая собственно ММП (25 генов), мембранно–связанные ММП, ADAM протеиназы (дизинтегрин–металлопротеиназные домены), ADAMTS протеиназы (дизинтегрин–металлопротеиназные домены с тромбоспондиновым мотивом) и ряд других.
Несбалансированный протеолиз компонентов ВКМ, порождаемый избыточной активностью ММП, был связан с рядом заболеваний (в том числе, артрит, рак, атеросклероз, аневризм аорты и фиброз) [27]. Активность индивидуальных ММП может регулироваться взаимодействиями со специфическими ингибиторами тканевых металлопротеиназ (TIMP белки). Для каждой специфической ММП существует специфический TIMP белок (например, для MMP1 ингибитор TIMP1 и т.д.). Специфические ММП, которые деградируют коллагеновые волокна, таким образом удаляя основные структурные опоры соединительной ткани известны под названием коллагенaз (табл. 1). Важно отметить, что практически все внемембранные ММП характеризуются весьма сход ной полноатомной структурой индивидуальных глобул, и каждая глобула фермента включает четыре обязательных Ca2+ и два Zn2+ иона (рис. 3).
Таблица 1. Коллагеназы и их субстраты
| Ген | Название фермента | Функция |
|---|---|---|
| MMP1 | Фибробласт коллагенaза | Деградирует коллагены I, II, III |
| MMP2 | Желатиназа А | Деградирует коллаген IV; активируется мембранно–связанными ММП (MMP14, MMP16, MMP17, MMP24, MMP25) |
| MMP3 | Прожелатиназа (син. стромелизин) | Деградирует коллагены III, IV, IX и X, а также фибронектин, ламинины и протеогликаны |
| MMP8 | Нейтрофил коллагеназа | Деградирует коллагены I, II, III |
| MMP9 | Макрофаг желатиназа | Деградирует коллагены IV, V |
| MMP13 | Коллагеназа 3 | Деградирует коллаген II и в меньшей степени коллагены I и III |
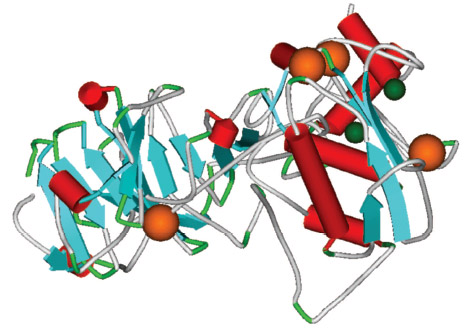
Рис. 3. Трехмерная структура профермента MMP1 человека.
Глобула (PDB код 1su3) состоит из двух структурных доменов, N–концевого каталитического (справа) и C–концевого регулирующего (слева). С–концевой домен определяет субстратную специфичность протеиназы, а также взаимодействует с TIMP белками (ингибиторами ММП). Четыре иона Ca2+ (большие сферы) и два иона Zn2+ (малые темные сферы) необходимы для катализа и стабилизации глобулы ММП.
Рисунок был сделан с помощью программы Weblab Viewer (msi.com)
Эластиновые волокна ВКМ придают эластичность внеклеточной матрице и соединительной ткани. Основной компонент этих волокон – эластин. Эластин составляет приблизительно 50% сухого веса артерий. В отличие от коллагенов эластин представлен только одним геном (ELN на хромосоме 7). Мутации данного гена приводят к стенозу аорты [28] и других артерий в результате чрезмерного количества клеток гладкой мышечной ткани в стенках артерий.
В отличие от коллагенов полипептидные цепи эластина не гликозилируются и не содержат гидроксилизинов. После синтеза на рибосоме и гидроксилирования пролиновых остатков прекурсор эластина секретируется в ВКМ, где он проходит через самосборку и поперечную сшивку между лизиновыми остатками [14]. Гидроксилирование пролинов и поперечная сшивка выполняются через те же биохимические пути, что и для коллагенов.
Гибкие эластиновые волокна не сформированы исключительно лишь эластином. Состоящая из эластиновых цепей сердцевина эластиновых волокон защищена снаружи гликобелками микрофибрил, которые включают фибриллины (гены FBN1, FBN3), фибулины (гены FBLN1, FBLN2, FBLN5) и эмилины (EMILIN1, EMILIN2, EMILIN3, EMILIN4). Эти сравнительно мало исследованные белки регулируют интерфейс между эластиновой сердцевиной и микрофибриллами, а также позволяют осуществлять тонкую подстройку эластичности волокон. Мыши с делецией гена EMILIN1 имеют повышенное кровяное давление вследствие возрастающего сопротивления в периферийной сосудистой системе и суженного просвета сосудов [29].
Гибкие волокна ремоделируются (деградируются) эластазами (гены ELA2A, ELA2B ELA3B, ELA3A, ELA1, ELA2). Фактически некоторые из ММП – также эластазы (например, фермент MMP12 известен, как «эластаза макро бактериофагов»).
Таким образом, представленное выше краткое описание функций белков внеклеточной матрицы позволяет сформулировать многочисленные молекулярные механизмы, через которые могла бы возникнуть дисплазия соединительной ткани. Любой дисбаланс в этой тонко на строенной системе соединительной ткани, будь то аномальная пролиферация ткани (вследствие, например, генетических дефектов в гене TGFBR1 рецептора TGF–β [30]), избыточная деградация коллагенoв, дефекты в структурных генах (протеогликанах, коллагенах, эластинах и т.д.) или аномалии в посттрансляционных модификациях, может приводить к ДСТ. Для того, чтобы ограничиться механизмами, которые связывают именно дефицит магния и этиологию ДСТ, мы проанализируем молекулярные механизмы гомеостаза магния, а затем представим синтез имеющихся данных.
Молекулярные механизмы гомеостаза магния
Среди катионов, присутствующих в организме человека, ион магния (Mg2+) находится на четвертом месте по распространенности (после натрия, калия и кальция). Mg2+ существенен для адгезии и миграции клеток, энергетического метаболизма, транскрипции ДНК, стабильности РНК, белкового синтеза, а также других клеточных функций [31–33].
В аннотированной последовательности генома человека существует не менее 290 генов и белковых продуктов, о которых известно, что они связывают Mg2+ как кофактор. Так как в геноме человека известна функция менее чем половины всех генов (14000 из 31000), можно предположить, что число магний–зависимых белков будет превышать 500. Так что на молекулярном уровне спектр влияния магния на физиологию человека едва ли может быть описан в нескольких словах.
С физиологической точки зрения до 53% магния концентрируется в костной ткани, дентине и эмали зубов и около 20% – в тканях с высокой метаболической активностью (мозг, сердце, мышцы, надпочечники, почки, печень). Только 10% всего магния в организме человека находится вне клеток, и 90% магниевых ионов концентрируются внутри клеток в форме Mg2+ ATФ (30% в митохондриях, 50% в цитозоле и 10% в ядре). У здорового человека концентрация магния в сыворотке крови поддерживается в достаточно узком диапазоне (норма 0,7–1,1 ммоль/л). Этот внеклеточный магний находится в непрерывном обмене с магниевыми запасами костей и мышечной ткани. Сбалансированная диета должна содержать ≈400 мг магния в сутки, из которого адсорбируется, как правило, около 200 мг.
Уменьшение количества ежедневно принимаемого магния может компенсироваться возрастающей адсорбцией магния в кишечнике и уменьшением выделения его через почки. Эти процессы транспорта Mg2+ регулируются рядом гормонов, включая антидиуретический пептид, глюкагон, кальцитонин, гормон паращитовидной железы (паратгормон) и инсулин [34]. Дефицит магния может сопровождаться вторичными ион–дефицитами, включая гипокалиемию, гипофосфатемию и гипокальциемию. Хронический дефицит магния может приводить к анорексии, тошноте и периодической слабости, к общему снижению тонуса мускулатуры, тахикардии, судорогам в мышцах, резко выраженной астенизации, вплоть до формирования синдрома хронической усталости [35].
Физиологически гипомагнезия может быть следствием уменьшенного приема пищевого магния, плохой адсорбции в тонком кишечнике или увеличения выделения через почки. Дефицит магния может быть генетически обусловленным или быть связанным с внешними факторами (несбалансированное питание, хронический эмоциональный стресс). В общих чертах, известные генетические заболевания, приводящие к отчетливой гипомагнезии, сравнительно редки (1:50000). Тем не менее гипомагнезия часто обнаруживается у пациентов с сахарным диабетом 2 типа, артериальной гипертонией, ИБС, бронхиальной астмой. Обострение дефицита магния согласуется с ухудшением протекания этих заболеваний и развитием осложнений [35,36].
Биодоступность магния в организме регулируется рядом генов, среди которых TRPM6 и TRPM7 наиболее важны. Белок TRPM6 (англ. «transient receptor potential cation channel 6») является ионным каналом, транспортирующим двухвалентные катионы. TRPM6 специфически взаимодействует с другим Mg2+–проницаемым каналом TRPM7, что приводит к сборке функциональных TRPM6/TRPM7 комплексов на поверхности клетки [37]. Мутации в TRPM6 могут приводить к гипомагнезии и вторичной гипокальциемии [38]. TRPM7 может быть во влечен в дефицит магния, связанный с эмоциональным стрессом под действием катехоламинов [39].
При гипомагнезии увеличивается экспрессия и другого гена ионного транспортера SLC41A1 (сольват–транс портер типа 41.1): у мышей на безмагниевой диете экспрессия мРНК гена SLC41A1 увеличивается в почках, кишечнике и сердце. В дополнение к Mg2+, SLC41A1 также может транспортировать Sr2+, Zn2+, Cu2+ и Co2+ [40]. Небольшие мембранные белки, включающие в себя FXYD–домены (домены, содержащие специфический фрагмент аминокислотной последовательности – фенилаланин–X–тирозин–аспартат), также вовлечены в транспорт магния. Мутации аминокислотных остатков в белке FXYD2 приводили к почечной гипомагнезии [41,42].
Клаудины (гены CLDN16 и CLDN19) – трансмембранные белки, обнаруженные на плотных соединениях клеток. Плотные соединения клеток (англ. «tight junction») образуют барьеры, которые контролируют транспорт ионов и метаболитов поперек листа эпителиальных клеток, а также перемещение белков и липидов между апикальными и базолатеральными областями эпителиальных клеток [43]. Ген CLDN16 (парацеллин 1) экспрессируется на плотных соединениях почечных эпителиальных клеток, находящихся на толстом восходящем отростке петли Генле, и играет центральную роль в реабсорбции двухвалентных катионов. Генетические дефекты в клаудине–16 были связаны с первичной гипомагнезией [44], а дефекты клаудина–19 приводят к почечной гипомагнезии с вовлечением глазной симптоматики (миопия, подвывих хрусталика) [45].
Регулирует обмен магния и Ca2+/Mg2+–чувствительный рецептор (ген CASR) – G–белок–связанный рецептор плазматической мембраны, который экспрессируется в паращитовидной железе и в почечных канальцах. Благо даря высокой чувствительности к небольшим изменениям в концентрациях циркулирующих кальция и магния CASR действует как сенсор (датчик), реагирующий на концентрацию катионов, и играет существенную роль в поддержании катионного гомеостаза [46]. Дефекты в этом гене связаны как с гиперкальциемией, так и с гипокальциемией [47]. Активация гена CASR Ca2+/Mg2+–чувствительного рецептора уменьшает активность белковой киназы А (PKA). Это приводит к уменьшению фосфорилирования клаудина–16, транслокации клаудина–16 в лизосомы, а в результате – к уменьшению реабсорбции магния в почечных канальцах [48].
Металлотионенин 2A (MT2A), возможно, играет важную роль в защите клеток от воспалительных реакций [49]. В физиологических условиях МТ2А вовлечен в регулирование гомеостаза цинка [50]. Аллель G полиморфизма +838 C/G соответствует повышению белка острой фазы MCP1 и уменьшению уровней цинка, меди и магния в эритроцитах, возрастанию уровня железа в плазме, а также повышенной склонности к образованию мягких атеросклеротических бляшек [51].
Возможные роли магния в метаболизме соединительной ткани
Точные и разносторонние гистологические данные о дисплазии соединительной ткани были бы весьма полезны для рассмотрения наиболее вероятных молекулярных механизмов болезни. Увы, тщательных исследований гистологии нДСТ не проводилось, так что характерные изменения морфологии соединительной ткани, связанные именно с нДСТ, неизвестны. Не имеется также гистологической характеристики соединительной ткани при дефиците магния, хотя и известно, что гипомагнезия приводит к изменению механических свойств артерий [52–54]. Мы можем допустить, что гистологические изменения при ги по магнезиях всегда будут ухудшать механические свойства соединительной ткани (прочность, эластичность) и на основе этого предположения рассмотреть различные молекулярные пути, через которые дефицит Mg2+ может отрицательно повлиять на структуру соединительной ткани.
Наиболее общий эффект воздействия Mg2+ на любую ткань заключается в том, что ионы Mg2+ необходимы для стабилизации некодирующих РНК. В частности, ион Mg2+ стабилизируют структуру транспортной РНК (рис. 4) и дефицит магния приведет к увеличению числа дисфункциональных молекул тРНК, таким образом снижая и замедляя общую скорость белкового синтеза.
Помимо стабилизации тРНК, Mg2+ также стабилизирует небольшие ядерные РНК (т.н. «snRNA»), которые участвуют в пре–сплайсинге кодирующей РНК на сплайсеосоме. Дефицит Mg2+ дестабилизирует snRNA и может приводить к частичной диссоциации (распаду) сплайсеосомы на РНК компоненты и белковые компоненты. Сыворотка пациентов с системными ревматическими болезнями часто содержит антитела против ядерных антигенов и среди них белки, образующие комплексы с snRNA (U1, U2, U4, U5 и U6 [55]). Результатом частичной диссоциации сплайсеосомы также будет замедление синтеза белка. Соответственно, дефицит магния в соединительной ткани приведет к замедлению синтеза всех структурных молекул (включая протеогликаны, глюкозаминогликаны, коллагены и эластин). По скольку синтез структурных молекул, столь необходимых для восстановления («ремонта») соединительной ткани, замедляется, то процессы восстановления также тормозятся, и это приводит к ухудшению механических характеристик ткани.
Совместное рассмотрение молекулярной биологии внеклеточной матрицы и физиологических механизмов гомеостаза магния может позволить нам сформулировать более специфические молекулярные механизмы, через которые может осуществляться взаимосвязь ДСТ и дефицита магния. Для того чтобы найти подобного рода взаимосвязи на уровне индивидуальных молекулярных генов, необходимо рассмотреть каждый из основных элементов внеклеточной матрицы в отдельности.
Основные элементы внеклеточной матрицы – это гелеобразная среда формируемая протеогликанами, структурными коллагеновыми волокнами, сетчатыми (ретикулярными) коллагеновыми волокнами, гибкими (эластиновыми) волокнами и клетками (фибробласты или хондробласты).
Никаких отчетливых эффектов Mg2+ на структуру гелеобразной субстанции ВКМ пока не было обнаружено. Все же возможно, что ионы Mg2+ могут модулировать активность соответствующих биосинтетических ферментов. Например, гиалуронансинтетазы HAS1, HAS2 и HAS3 содержат ион магния в активном центре (рис. 5). С другой стороны, известно, что действие ингибиторов гиалуронидаз (ферментов, деградирующих гиалуронан) зависит от концентрации ионов магния [56]. Таким образом, дефицит магния может приводить к понижению активности гиалуронансинтетаз и в то же время – к повышению активности гиалуронидаз (так как ингибиторы перестают действовать при недостатке магния). Оба эти процесса приведут к ухудшению механических свойств нитей гиалуронана и частичной деградации геля, образующего основу ВКМ.
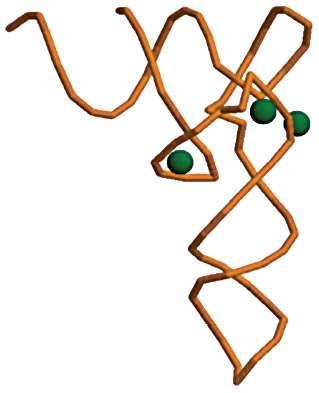 Рис. 4. Структура тРНК стабилизирована ионами магния (PDB 1I9V). Дефицит магния приводит к дестабилизации молекулы и частичной потере функциональных свойств
| 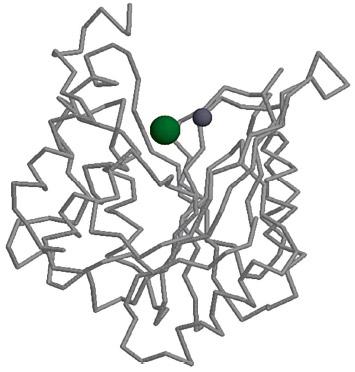 Рис. 5. Модель трехмерной структуры гиалуронансинтетазы 1. Показаны ион Mg2+ (большая, более темная сфера) и ион Mn2+ (меньшая сфера), размещенные в активном центре фермента. Модель создана на основе структуры полисахаридсинтетазы (PDB 1qgq) |
Коллагеновые волокна являются основной структурной поддержкой соединительной ткани. Избыток коллагеновых волокон или слишком малая активность коллагеназ приводит к увеличению плотности волокон и к формированию менее гибкой ткани. Наоборот, чрезмерная активность коллагеназ приведет к неуправляемой фрагментации коллагена, что сделает ткань более аморфной.
Непосредственно ионы Mg2+ не взаимодействуют ни с молекулами коллагена, ни с TIMP белками, так что эффект Mg2+ на активность коллагеназ особенно интересен. Известно, что лечение пациентов с острым инфарктом миокарда посредством сульфата магния и оротата магния тормозит повреждение миокарда. Обычно максимальные уровни IL6 и MMP1 в крови значительно возрастают при остром инфаркте миокарда, но эти уровни остаются на сравнительно низком уровне у пациентов после магний–терапии. Возрастающая концентрация IL6 может приводить к повышению общей активности MMP1, ведя к повреждению тканей, в то время как увеличение концентрации Mg2+ в сыворотке крови уменьшает уровни IL6 и MMP1 [57]. Дополнение диеты фолиевой кислотой и солями магния уменьшает MMP2 секрецию и оказывает положительное влияние на ИБС [58].
Эксперименты на животных подтверждают влияние магния на биологическую активность ММП. У мышей с искусственно вызванным дефицитом магния стенка аорты была значительно тоньше, чем у контрольных животных. Специфическая окраска двух основных типов волокон (коллагена и эластина) показала существенные структурные изменения обоих компонентов. Эти изменения кореллировали с повышением общей активности матричных металлопротеиназ MMP2 и MMP9 [59]. В клетках гладкой мускулатуры сосудов у крыс добавление магния уменьшало общую активность MMP2 прямо пропорционально дозе магния [60]. Возможно, что соответствующий сигнальный каскад запускается одним из белков–сенсоров магния (например, CASR, TRPM7, см. выше).
Данные, приведенные выше, позволяют предположить, что дефицит Mg2+ должен, вероятно, приводить к повышению активности ММП (в частности, коллагеназ), которые начинают деградировать структурные компоненты ВКМ (прежде всего коллаген) с более высокой скоростью. Эти эффекты воздействия магния, наиболее вероятно, вызываются через некий, пока не известный внутриклеточный сигнальный каскад. Нельзя исключать также и возможность прямого ингибирования различных ММП ионами магния через конкурентное связывание двухвалетных катионов в активном центре (рис. 5) или через аллостерические взаимодействия с ММП.
Эффекты Mg2+ на соединительную ткань не ограничиваются коллагеном и коллагенaзами. Микрофибрилы и эластин – основные компоненты гибких волокон. Деградация волокон эластина может значительно возрастать (в 2–3 раза) в присутствии Mg2+ [61]. Дефицит Mg2+ соответствует более низкой активности эластаз и большей концентрации гибких волокон. В основном, однако, скорее, кальциевые, а не магниевые ионы влияют на гибкие волокна: Ca2+ необходим для активных центров эластаз, Ca2+ также стабилизирует структуру микрофибрил (в частности, через взаимодействия с фибриллином–1 [62] и микрофибрил-связанным гликобелком–1 (MAGP–1) [63]).
Трансглутаминаза – фермент, формирующий поперечные глутамин-лизиновые сшивки, соединяющие вместе цепи эластина, активизируется Ca2+ и ингибируется Mg2+ [64,65]. Mg2+ может ингибировать медь–зависимую лизилоксидазу (LOX) [66], также вовлеченную в поперечную сшивку цепей эластинов и/или коллагенов. Соответственно, дефицит Mg2+ может приводить к активизации поперечной сшивки коллагенов и эластинов, и этот процесс, наряду с увеличением активности ММП, приведет к своего рода грануляризации соединительной ткани.
Таким образом, имеющиеся данные позволяют сделать вывод, что наиболее вероятные механизмы воздействия дефицита Mg2+ на соединительную ткань – это усиление деградации коллагеновых и, возможно, эластиновых волокон, а также полисахаридных нитей гиалуронана (рис. 6). Нельзя исключить, что усиление поперечных сшивок приведет к грануляризации соединительной ткани, расслоению на этакие «пластинки», состоящие из наполовину деградированных молекул коллагенов, и в результате приводит к уменьшению механической прочности. При достаточной концентрации Mg2+ секреция/активность ММП снижаются, что приводит к уменьшению деградации и к ускорению белкового синтеза новых молекул коллагенов.
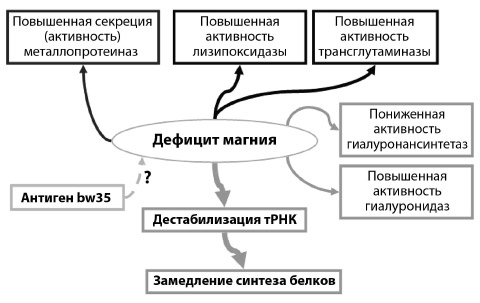
Рис. 6. Предлагаемые механизмы, связывающие дефицит магния со структурой соединительной ткани
Частный случай: Bw35 антиген (ген HLA–B)
Приведенный выше анализ отдельных компонентов внеклеточной матрицы предоставил возможность систематизировать взаимосвязь дефицита магния и метаболизма соединительной ткани. Существует, тем не менее, еще один конкретный случай, который может обусловливать ДСТ, не имеющий, однако, непосредственного от ношения к структуре ВКМ. Речь идет о край не интересных данных по антигену Bw35.
Известно, что пациенты с пролапсом митрального клапана часто характеризуются присутствием антигена Bw35 системы HLA (лимфоцитарный антиген гистосовместимости, часть MHC системы иммунной защиты) который таким образом (через факт присутствия заболевания) ассоциируется с абнормальным метаболизмом соединительной ткани [1,67] и с магний-дефицитом [1,68,69]. Аллель Bw35 системы HLA, наряду с поведенческим типом «А» (агрессивный, неуравновешенный тип поведения), представляет собой конституционный фактор, указывающий на повышенную потребность в магнии [70].
Рецептор MHC основного комплекса гистосовместимости типа 1 играет центральную роль в иммунной системе, презентируя пептид–производные белков цитозоля и эндоплазматического ретикулума на поверхности клеток. Такие рецепторы экспрессируются практически в каждой клетке, и T–лимфоциты атакуют те клетки, MHC рецепторы которых презентируют чужеродные пептиды. Возможно, что Т–лимфоциты будут атаковать клетки, содержащие определенные мутированные формы MHC рецепторов, которые признаются Т–лимфоцитами как «чужеродные». Этот процесс, очевидно, вызовет повышение аутоимунных реакций. Генетические дефекты гена HLA–B могут привести к спондилоартропатии, одному из наиболее распространенных хронических ревматических заболеваний, а также к другим сходным заболеваниям (например, анкилозному спондилиту [71,72]).
В частности, аллель bw35 гена HLA–B был ассоциирован с тяжелым протеканием гипертонии, тяжелым повреждением сосудов [73], псориазом [74], повышенной восприимчивостью к гепатиту B [75], миеломой [76], тироидитом [77], а также с другими аутоимунными заболеваниями [78], такими как воспалительная болезнь кишечника, ревматоидный артрит и прогрессирующая склеродермия. Все эти заболевания имеют воспалительный компонент, связанный с состоянием соединительной ткани хрящей/связок.
Вероятное молекулярное объяснение этому заключается в следующем: MHC рецепторы, соответствующие антигену bw35, провоцируют T–клетки атаковать ткани, содержащие эту форму рецептора. Это, в свою очередь, приводит к усиленной аутоимунной реакции и, как следствие, к усиленной деградации соединительной ткани. Повышенная деградация ткани через аутоимунный механизм неизбежно приведет к неконтролируемой потери магния из ткани и к увеличению выделения ионов магния через почки.
Другое возможное объяснение этого феномена включает хорошо известный в генетике эффект «неравновесного сцепления» (англ. «linkage disequilibrium»). Не равновесное сцепление участков хромосомы означает, что во время мейоза вероятность рекомбинации данных участков чрезвычайно мала и данные участки хромосомы наследуются единым блоком. В этом случае bw35 аллель не соответствует каким бы то ни было аномальным формам MHC рецептора, но выступает в качестве генетического маркера для генов, находящихся в неравновесном сцеплении с геном HLA–B. Как показывает совместный анализ последовательности и вариаций человеческого генома, у европеоидов кластер HLA генов (цитогенетическая полоса 6p21) вовлечен в очень большую группу неравновесного сцепления (более 2 Mb нуклеотидов). Эта группа сцепленных генов, которые наследуются, как единый блок у европеоидов, включает 14 генов. Среди них – три гена HLA–системы, гипотетический ген подобный дигидрофолатредуктазы (LOC729816), два гена, ассоциированных с восприимчивостью к псориазу (PSORS1C1, PSORS1C2), корнеодесмозин (CDSN), вовлеченный в десквамацию эпителия, и три гена, связанные с процессами дифференцирования и морфогенеза клеток (CCHCR1, TCF19, POU5F1). Функции остальных генов пока не известны. В данном случае bw35 антиген HLA–B может не быть функциональным аллелем, а просто являться генетическим маркером, указывающим на присутствие абнормальных форм любого из этих 14 генов.
Выводы
Вне зависимости от того, возникает ли аномалия в структуре соединительной ткани из–за аутоиммунного ответа, из–за абнормального метаболизма ткани и чрезмерной активности коллагеназ или же вследствие других причин, состояние соединительной ткани только улучшится, если активности коллагеназ и эластаз, а так же биосинтетических ферментов глюкозаминогликанов (гиалуронансинтетаз, гиалуронидаз, β–галактозидаз) будут сбалансированы.
Достижение такого баланса, по всей видимости, достигается непосредственным эффектом воздействия адекватных доз ионов магния. Напротив, при дефиците Mg2+ синтез белков в соединительной ткани замедляется, активность ММП увеличивается и внеклеточная матрица прогрессивно деградирует, так как структурная поддержка ткани (в частности, коллагеновые волокна) разрушается быстрее, чем синтезируется.
Анализ исследований, представленный в данной статье, позволил сформулировать ряд принципиально новых молекулярных механизмов взаимосвязи между Mg2+ и ДСТ. Наиболее вероятными механизмами являются следующие: 1) дестабилизации тРНК и сплайсеосом; 2) деактивация гиалуронансинтетаз и повышение активности гиалуронидаз; 3) активация матричных металлопротеиназ; 4) инактивация эластаз; 5) активация трансглутаминазы и лизилоксидазы, а также 6) аутоиммунные реакции, обусловленные аллелем bw35 гена HLA–B.
Нельзя исключить возможность того, что часто встречающиеся нуклеотидные полиморфизмы в соответствующих генах могут влиять на состояние соединительной ткани. Подтверждение сформулированных молекулярных механизмов позволит провести обоснованные медико-генетические исследования постгеномного характера [79]. Экспериментальные исследования предлагаемых молекулярных механизмов должны включать подробный анализ ультраструктурных изменений в CT, порождаемых Mg2+/Ca2+ дефицитом, гистологический и ультраструктурный анализ тканей пациентов с ДСТ, анализ взаимоотношений между активностью ММП и Mg2+ in vitro, в клетках в культуре а также in vivo и др.
Литература
Комментарии
ПРАКТИКА ПЕДИАТРА



