Asenapine versus olanzapine in people with persistent negative symptoms of schizophrenia
Статьи Опубликовано в журнале:«JOURNAL OF CLINICAL PSYCHOPHARMACOLOGY»; Volume 32; № 1; Февраль 2012; стр. 36-45.
Robert W. Buchanan, MD,* John Panagides, PhD,† Jun Zhao, PhD,‡ Phillip Phiri, PhD,‡ Wil den Hollander, MSc,‡ Xianwei Ha, PhD,‡ Alex Kouassi, PhD,‡ Larry Alphs, MD, PhD,§ Nina Schooler, PhD,?? Armin Szegedi, MD, PhD,‡ and Pilar Cazorla, PhD‡
Abstract: Two randomized, double-blind, 26-week core studies (Eastern [EH] and Western Hemisphere [WH]) tested the hypothesis that asenapine is superior to olanzapine for persistent negative symptoms of schizophrenia; 26-week extension studies assessed the comparative longterm efficacy and safety of these agents. In the core studies, 949 people were randomized to asenapine (n = 241 and 244) or olanzapine (n = 240 and 224); 26-week completion rates with asenapine were 64.7% and 49.6% (olanzapine, 80.4% and 63.8%) in the EH and WH, respectively. In the EH and WH extensions, respectively (asenapine, n = 134 and 86; olanzapine, n = 172 and 110), 52-week completion rates were 84.3% and 66.3% with asenapine (olanzapine, 89.0% and 80.9%). Asenapine was not superior to olanzapine in change in the 16-item Negative Symptom Assessment Scale total score in either core study, but asenapine was superior to olanzapine at week 52 in the WH extension study. Olanzapine was associated with modest, but significantly greater, changes in PANSS positive subscale score at various assessment times in both core studies and the WH extension study. Incidence of treatment-emergent adverse events was comparable between treatments across studies. Weight gain was consistently lower with asenapine. Extrapyramidal symptom-related adverse event incidence was higher with asenapine (EH: 8.3%; 95% confidence interval [CI], 5.1%-12.5%; WH: 16.4%; 95% CI, 11.9%-21.6%) than olanzapine (EH: 3.3%; 95% CI, 1.4%-6.4%; WH: 12.1%; 95% CI, 8.1%-17.0%), but Extrapyramidal Symptom Rating Scale-Abbreviated total score changes did not significantly differ between treatments. In conclusion, asenapine superiority over olanzapine was not observed in the core studies. Both treatments improved persistent negative symptoms, but discontinuation rates were higher with asenapine.
Key Words: asenapine, NSA-16, olanzapine, persistent negative symptoms, schizophrenia
Negative symptoms represent a key symptom domain in schizophrenia and substantially contribute to long-term morbidity and poor outcome in people with schizophrenia.1'2 Persistent negative symptoms (PNS), either primary to the illness or a treatment-resistant secondary problem, persist during periods of clinical stability.3 Although PNS represent a critically important unmet therapeutic need,4,5 there has been scant research involving large-scale psychopharmacology trials for this symptom domain.3
In a previous 6-week, randomized, placebo-controlled comparison of asenapine and risperidone, both drugs were superior to placebo in treating the positive symptoms of schizophrenia, but only asenapine demonstrated superiority over placebo in treating negative symptoms.6 The present report describes the results of 4 long-term studies (two 26-week core studies each followed by a 26-week extension) designed to compare the effects of asenapine and olanzapine in people with schizophrenia selected for PNS. The 2 core studies were designed to test the hypothesis that asenapine is superior to olanzapine for the treatment of PNS; the 2 extension studies were designed to assess long-term safety and the continued efficacy of asenapine and olanzapine for PNS. The PNS outcome is designed to maximize the likelihood that participants will have primary negative symptoms, will not have secondary negative symptoms that are associated with an acute exacerbation of the illness, and will facilitate methodologically well-designed pharmacological research into a clinical problem of substantial magnitude and relevance.3,7
Materials and methods
Study Design
Two sets of randomized double-blind studies were conducted between May 2005 and May 2009; each set consisted of a 26-week core study and a 26-week extension. The Eastern Hemisphere studies (EH: 25543 and 25544; clinical trials registry identifiers NCT00212836 and NCT00265343, respectively) were conducted in 15 countries (72 centers) in Europe, South Africa, and Australia (Supplementary Fig. A, Supplemental Digital Content 1). The Western Hemisphere studies (WH: A7501013 and A7501014; NCT00145496 and NCT00174265) were conducted in 5 countries (95 centers) in North and South America (Supplementary Fig. A, Supplemental Digital Content 1). These studies were identical in design, except for a few minor differences in assessments, and were conducted in accordance with principles of Good Clinical Practice and were approved by the appropriate institutional review boards and regulatory agencies.
Inclusion and Exclusion Criteria
Core Studies
Men and women 18 years or older (primarily outpatients) meeting Diagnostic and Statistical Manual of Mental Disorders, 4th Edition, Text Revision criteria for schizophrenia were eligible for participation; schizophrenia was diagnosed using the Mini International Neuropsychiatric Interview.8 All subjects provided written informed consent after the study procedures had been fully explained and before study participation; if consent was obtained from the legal representative, subjects provided assent to their participation. In addition, all subjects had to be fluent in the language of the investigative team and to have identified a responsible person to support compliance with study procedures.
Eligible subjects had a Positive and Negative Syndrome Scale (PANSS) negative symptom subscale9 score of 20 or greater at screening and baseline, and scores of 4 or greater (moderate) on 3 or more of the 7 PANSS items of the negative symptom factor (blunted affect, emotional withdrawal, poor rapport, passive social withdrawal, lack of spontaneity, motor retardation, active social avoidance) of Marder et al.10 In addition, subjects had to be clinically stable for 5 months before screening (ie, no antipsychotic dosage increases or changes in medication to treat symptoms of schizophrenia, no hospitalizations or increases in level of psychiatric care, no imprisonment related to worsening of schizophrenia symptoms). Women of childbearing potential had to be nonpregnant, nonlactating, and using an acceptable method of birth control.
Subjects were excluded if they had an Extrapyramidal Symptom Rating Scale-Abbreviated (ESRS-A)11 global Parkinson item score of 3 or greater, a Calgary Depression Scale for Schizophrenia (CDSS)12 total score of 9 or greater, or a rating of 4 or greater on 2 or more PANSS positive symptom subscale items. Subjects were also excluded if they had a clinically significant medical condition that could interfere with the interpretation of safety or efficacy evaluations in the opinion of the investigator; anorexia (body mass index [BMI] of less than 18.5 kg/m2) or obesity (BMI, >35 kg/m2); clinically significant abnormalities in laboratories, vital signs, physical examinations, or electrocardiograms at screening; psychosis or behavioral disturbance attributed to substance abuse; current (?6 months before screening) Diagnostic and Statistical Manual of Mental Disorders, 4th Edition, Text Revision diagnosis of substance abuse/dependence disorder (excluding nicotine); concurrent Axis I psychiatric disorders other than schizophrenia; or mental retardation or severe organic brain syndrome or were an imminent risk of harm to themselves or others.
Furthermore, subjects were excluded if they were treated with olanzapine within 5 months of screening and had an inadequate negative symptom response despite adequate dosages of olanzapine for 3 months or greater, had been treated with clozapine within 5 months of screening for treatment-resistant schizophrenia, had a history of hypersensitivity to olanzapine, received antidepressants and/or mood stabilizers to treat a major depressive disorder or had antidepressant dosage changes due to clinically unstable depressive symptoms within 5 months of screening, previously participated in an asenapine trial, used an investigational drug within 30 days of baseline, required high doses of benzodiazepines to maintain clinical stability (?4 mg/d lorazepam or equivalent), or were judged by the investigator to be medically noncompliant in the management of their disease.
Subjects were required to demonstrate continued stability, which was defined as no screening to baseline change in Clinical Global Impression-Severity of Illness (CGI-S) score13 and screening to baseline changes in PANSS total score and PANSS Marder factor negative symptom score within ±20% of baseline. Subjects who met these criteria were eligible for randomization.
Extension Studies
Subjects completing either of the core studies with acceptable medication compliance were eligible to participate if in the investigator's judgment continued treatment would be beneficial and if written informed consent was provided by the subject or a legally authorized representative.
Study Treatments
In the core studies, subjects satisfying screening criteria entered a 30-day observation period to establish continued clinical stability. During this period, use of prestudy antipsychotic medications continued until the double-blind treatment phase began. Subjects who continued to meet all inclusion criteria entered the 26-week double-blind treatment phase and were randomized to asenapine or olanzapine. Those randomized to asenapine received sublingual asenapine 5 mg twice a day for 1 week, with subsequent dose adjustments between 5 and 10 mg twice a day at the discretion of the investigator, and placebo olanzapine oral tablets. Subjects randomized to olanzapine received olanzapine oral tablets 10 mg once daily for 1 week, with subsequent dose adjustments between 5 and 20 mg once daily at the discretion of the investigator, and sublingual dissolving placebo asenapine tablets. The stratification variable for randomization was duration of PNS (<2 vs ?2 years).
Subjects were tapered off of their prestudy antipsychotics during the first 4 weeks of double-blind treatment. Subjects could continue to receive benzodiazepine treatment for agitation/anxiety (<4 mg/d lorazepam or equivalent) and partial benzodiazepine agonists for insomnia/sleep disturbance. Concomitant medication for extrapyramidal symptoms (EPS) was gradually tapered from days 7 to 28 based on the investigators' judgment and was then discontinued unless there was a documented requirement for continued treatment. If EPS reappeared, concomitant EPS medication was permitted and further ESRS-A assessments were performed.
Subjects entering the 26-week double-blind extension studies continued on their core study treatment regimens without rerandomization. Dosage adjustment was allowed as needed within the same limits as applied during the core study.
Efficacy Assessments
The 16-item Negative Symptom Assessment Scale (NSA-16) total score14 was the primary efficacy variable used to assess negative symptoms. The PANSS total and subscale scores, the PANSS Marder factor scores (positive, negative, disorganized thought, hostility/excitement, anxiety/depression), the CGI-S and CGI-Improvement (CGI-I) scales,13 the CDSS total score,12 and the Quality of Life Scale (QLS)15 total score were secondary efficacy variables. Efficacy assessments were conducted at screening, baseline, and weeks 1, 2, 3, 4, 6, 8, 12, 16, 20, and 26 in the core and every 4 to 6 weeks in the extension studies.
Safety Assessments
Clinical laboratory testing was performed at screening, baseline, and weeks 4, 12, and 26 in the core and at weeks 38 and 52 in the extension studies; vital sign measurements were performed at screening, baseline, and weeks 1, 4, 8, 12, 16, 20, and 26 in the core and every 4 to 6 weeks in the extension studies; physical examinations were conducted at screening and weeks 12 and 26 in the core and at weeks 38 and 52 in the extension studies; and electrocardiograms were obtained at screening, baseline, and weeks 6,16, and 26 in the core studies and at weeks 38 and 52 in the extensions.
All reported adverse events (AEs), serious AEs (SAEs), and treatment discontinuations were recorded and coded using the Medical Dictionary for Regulatory Activities (version 10.0, 11.1, or 12.0). The ESRS-A was performed at regular study visits, when EPS worsened, or when there was any change in EPS medication use. The percentage of subjects meeting the National Cholesterol Education Program (NCEP)16 criteria for metabolic syndrome was calculated at baseline and endpoint.
Rater Training
Raters were required to meet prespecified qualifications and to exhibit proficiency on key scales before performing any ratings. Proficiency on the PANSS and NSA-16 was evaluated through the use of videotaped interviews, with raters required to be within 1 point of the modal rating on 80% of the items or greater on each scale. They were also required to maintain this prespecified level of interrater reliability throughout the course of the study through use of online recertification modules for the PANSS and NSA-16.
Statistical Analysis
Efficacy and safety analyses were based on the changes from baseline of the core study to week 26 or end point of the core study and on changes from baseline of the core study to week 52 or end point of the extension studies. Extension data included only subjects entering the extension studies, with safety analyses being based on data from both the core and extension studies (ie, 1 year of exposure).
The core studies used an adaptive design17 in which an interim analysis was conducted after 120 subjects had NSA-16 (primary end point) and QLS ratings (key secondary end point) at week 26 or had withdrawn from the study; this analysis was used to determine if an increase in the sample size was required. Interim data were analyzed and reviewed by an independent external interim analysis review board, who recommended against an increase in sample size.
A mixed model for repeated measures (MMRM) was used for both the core and extension studies. The MMRM used change from baseline in NSA-16 total score at each visit as the dependent variable and included factors of treatment, pooled center (small centers were pooled to form pseudocenters), visit, treatment-by-visit interaction, and duration of PNS, with baseline NSA-16 total score included as a covariate. The variance-covariance matrix of the repeated measurements was specified as unstructured in the model. The treatment-by-center interaction was also explored. The comparison of asenapine with olanzapine on the change in NSA-16 total scores was performed using a 2-tailed test, with a significance level of P < 0.05. The robustness of the primary results was checked with analysis of covariance (ANCOVA) using last observation carried forward (LOCF) and with an observed case (OC) analysis. These models included factors of treatment, pooled center, and duration of PNS; baseline value was included as a covariate.
Selected secondary outcomes (eg, change from baseline in the PANSS Marder factors, PANSS subscale scores, and CGI-S scores) and weight changes were also analyzed using an MMRM and ANCOVA with LOCF. Other tertiary end points (eg, change from baseline in the CDSS scores) were analyzed using ANCOVA with LOCF. Subjects classified as ''very much improved'' and ''much improved'' on the CGI-I were defined as CGI-I responders. CGI-I responder rates in each group were compared using the Cochran-Mantel-Haenszel (CMH) ?2 test.
Descriptive statistics were used to assess treatment discontinuation rates, AEs, changes in ESRS-A, and changes in laboratory and clinical assessments. Between-group differences in percentages of subjects with metabolic syndrome at end point and baseline-to-end point shift in metabolic syndrome classification were compared using the CMH test adjusted by site. All analyses were conducted using SAS version 9.1 (Cary, NC).
Results
Subject Disposition and Group Characteristics at Baseline
In the core studies, 949 people were randomized to asenapine (EH: n = 241 and WH: n = 244) or olanzapine (n = 240 and n = 224, respectively); 26-week completion rates were 64.7% and 49.6% with asenapine and 80.4% and 63.8% with olanzapine in the EH and WH studies, respectively. In the EH and WH extension study populations, respectively (asenapine, n = 134 and 86; olanzapine, n = 172 and 110), 52-week completion rates were 84.3% and 66.3% with asenapine and 89.0% and 80.9% with olanzapine (Supplementary Fig. A, Supplemental Digital Content 1).
There were no significant between-group differences in baseline demographic or clinical characteristics among subjects entering double-blind treatment in the core studies or the extension studies (Supplementary Table A, Supplemental Digital Content 2). The rate of antipsychotic use before randomization was similar for asenapine versus olanzapine in the EH (96.3% vs 95.8%) and WH (93.9% vs 96.9%) studies, with second-generation antipsychotics being used most often. Antipsychotics used in 10% or more of randomized subjects in any group (asenapine vs olanzapine) included risperidone (EH: 27.0% vs 27.5%; WH: 36.5% vs 33.9%), olanzapine (EH: 23.7% vs 16.7%; WH: 24.6% vs 24.6%), quetiapine (EH: 7.1% vs 9.6%; WH: 14.3% vs 13.4%), haloperidol (EH: 13.7% vs 10.0%; WH: 9.0% vs 11.6%), and aripiprazole (EH: 2.9% vs 6.7%; WH: 7.4% vs 9.8%).
In both core studies, discontinuation rates were higher with asenapine than olanzapine (EH: 35.3% vs 19.6%; WH: 50.4% vs 36.2%), as was the occurrence of AEs involving worsening symptoms as a cause for discontinuation. In the EH study, protocol violations (eg, data irregularities, lack of postbaseline NSA-16 assessment, enrollment in a previous asenapine study) resulted in the exclusion of 48 subjects. During the extension studies, discontinuation rates were higher in the WH than in the EH and were higher with asenapine than olanzapine in both studies (EH: 15.7% vs 11.0%; WH: 33.7% vs 19.1%; Supplementary Fig. A, Supplemental Digital Content 1).
Dosing and Concomitant Medication
In the EH core study, daily dose of study medication (mean ± SD) was as follows: asenapine, 14.4 ± 4.1 mg; olanzapine, 12.5 ± 4.2 mg; in the WH core study: asenapine, 14.5 ± 4.4 mg; olanzapine, 14.0 ± 4.0 mg; in the EH extension: asenapine, 15.9 ± 4.7 mg; olanzapine, 12.8 ± 5.2 mg; in the WH extension: asenapine, 16.0 ± 4.3 mg; olanzapine, 14.8 ± 4.3 mg. Compliance rates for both drugs (based on the number of doses observed in the returned packages and subject interviews) ranged from 75% to 125% across all studies.
During the postrandomization 28-day tapering period, continued use of pretrial antipsychotics was lower in the EH studies than in the WH studies (Supplementary Table B, Supplemental Digital Content 3). The use of antidepressants and mood stabilizers up to day 28 was slightly higher in the EH compared with the WH studies and was also higher in the EH studies after day 28. Post hoc CMH analysis found no significant group differences in use of antipsychotics, mood stabilizers, or antidepressants up to day 28 or after day 28 in any of the studies.
The use of antiparkinson medications tended to be lower in the EH than in the WH (core, 16.6% vs 25.0%%; extensions, 20.3% vs 26.7%; Supplementary Table B, Supplemental Digital Content 3), as was the percentage of subjects initiating usage after day 7 (core, 1.5% vs 8.1%; extensions, 1.0% vs 7.7%). Post hoc CMH analyses indicated that between-group differences were not statistically significant in antiparkinson medication use.
Efficacy
Core Studies
Although both treatments reduced negative symptoms, there were no significant between-group differences on the primary outcome measure, change from baseline NSA-16 total score at week 26 (EH: P = 0.79; WH: P = 0.72; Fig. 1, A and B; Table 1). In both studies, change in NSA-16 total score was similar in subjects with PNS duration of less than 2 years versus 2 years or greater. Results with LOCF and OC analyses were consistent with these MMRM analyses.
FIGURE 1. LS mean ± SE changes from baseline in NSA-16 total scores during 26 weeks of treatment in the core studies using MMRM analysis (A and B in the EH and WH regions, respectively) and in PANSS positive subscale scores (C and D) and CDSS total scores (E and F) using LOCF analyses. Change from baseline on NSA-16 total score was the primary end point; all other end points were secondary. ASEN indicates asenapine; OLAN, olanzapine. A and B, Based on a mixed model with defined covariance structure (unstructured), treatment, and pooled investigative site as fixed effects and baseline and duration of persistent negative symptom as covariates. C-F, Based on an ANCOVA model with treatment and pooled investigative site as fixed effects, baseline and duration of predominant negative symptom as covariates. 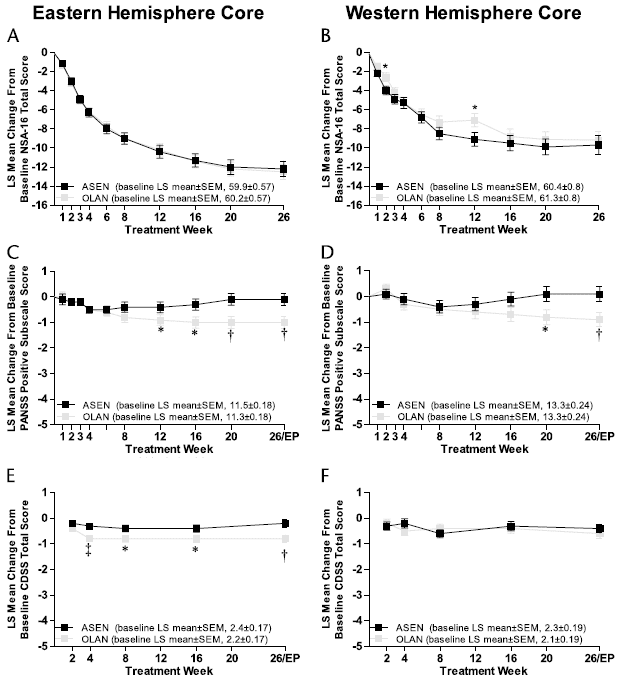
TABLE 1.
Changes From Baseline on Efficacy Measures During the Core Studies (LS Mean ± SE), Intent to Treat
|
|
|
Changes From Baseline | |||||
|
Baseline | Week 4 | Week 8 | Week 16 | Week 26 | |||
| NSA-16 total score* EH | ASEN | 59.9 ± 0.57 | -6.3 ± 0.45 | -9.0 ± 0.59 | -11.3 ± 0.70 | -12.2 ± 0.81 | |
| OLAN | 60.2 ± 0.57 | -6.2 ± 0.44 | -9.0 ± 0.57 | -11.4 ± 0.67 | -12.5 ± 0.76 | ||
| WH | ASEN | 60.4 ± 0.80 | -5.3 ± 0.58 | -8.5 ± 0.66 | -9.5 ± 0.80 | -9.7 ± 0.95 | |
| OLAN | 61.3 ± 0.80 | -5.3 ± 0.59 | -7.3 ± 0.65 | -8.8 ± 0.77 | -9.2 ± 0.89 | ||
| QLS* EH | ASEN | 46.4 ± 1.02 | NA | 7.5 ± 0.86 | 11.1 ± 1.01 | 11.7 ± 1.14 | |
| OLAN | 47.1 ± 1.01 | NA | 6.7 ± 0.81 | 9.1 ± 0.94 | 11.8 ± 1.05 | ||
| WH | ASEN | 46.3 ± 1.20 | NA | 6.8 ± 1.06 | 8.6 ± 1.25 | 11.1 ± 1.54 | |
| OLAN | 44.2 ± 1.20 | NA | 4.9 ± 1.00 | 6.1 ± 1.19 | 7.1 ± 1.41 | ||
| PANSS negative subscale* EH | ASEN | 27.1 ± 0.25 | -3.2 ± 0.22 | -4.7 ± 0.28 | -6.2 ± 0.33 | -7.1 ± 0.38 | |
| OLAN | 26.7 ± 0.25 | -2.9 ± 0.22 | -4.3 ± 0.27 | -5.8 ± 0.32 | -6.6 ± 0.35 | ||
| WH | ASEN | 26.7 ± 0.33 | -3.7 ± 0.27 | -4.9 ± 0.31 | -6.0 ± 0.37 | -6.3 ± 0.48 | |
| OLAN | 27.1 ± 0.33 | -3.9 ± 0.27 | -4.9 ± 0.30 | -5.7 ± 0.36 | -6.5 ± 0.44 | ||
| PANSS Marder factor for negative symptoms* EH | ASEN | 26.9 ± 0.25 | -3.8 ± 0.25 | -5.5 ± 0.29 | -7.2 ± 0.36 | -8.0 ± 0.40 | |
| OLAN | 26.7 ± 0.25 | -3.5 ± 0.25 | -5.0 ± 0.29 | -6.6 ± 0.35 | -7.4 ± 0.37 | ||
| WH | ASEN | 26.8 ± 0.34 | -4.1 ± 0.29 | -5.6 ± 0.32 | -6.7 ± 0.39 | -7.0 ± 0.48 | |
| OLAN | 26.9 ± 0.34 | -4.1 ± 0.29 | -4.9 ± 0.31 | -5.7 ± 0.37 | -6.7 ± 0.45 | ||
| PANSS total score* EH | ASEN | 74.7 ± 0.66 | -7.0 ± 0.51 | -9.6 ± 0.74 | -12.4 ± 0.85 | -13.6 ± 0.93 | |
| OLAN | 73.8 ± 0.66 | -7.0 ± 0.51 | -9.7 ± 0.73 | -12.7 ± 0.81 | -14.2 ± 0.87 | ||
| WH | ASEN | 76.3 ± 0.86 | -6.6 ± 0.69 | - 10.1 ± 0.78 | -10.7 ± 0.93 | -11.6 ± 1.14 | |
| OLAN | 75.8 ± 0.86 | -7.6 ± 0.69 | -9.3 ± 0.76 | -11.0 ± 0.90 | -13.8 ± 1.07 | ||
| PANSS positive subscale† EH | ASEN | 11.5 ± 0.18 | -0.5 ± 0.13 | -0.4 ± 0.20 | -0.3 ± 0.22 | -0.1 ± 0.23 | |
| OLAN | 11.3 ± 0.18 | -0.5 ± 0.13 | -0.8 ± 0.20 | -1.0 ± 0.22 | -1.0 ± 0.23 | ||
| WH | ASEN | 13.3 ± 0.24 | -0.1 ± 0.22 | -0.4 ± 0.24 | -0.1 ± 0.27 | 0.1 ± 0.28 | |
| OLAN | 13.3 ± 0.24 | -0.3 ± 0.22 | -0.5 ± 0.24 | -0.7 ± 0.27 | -0.9 ± 0.28 | ||
*Based on an MMRM with covariance structure set as unstructured, treatment, pooled investigative site, visit, treatment by visit interaction, and duration of predominant negative symptoms as fixed effects, and baseline as covariate.
†Based on an ANCOVA model with treatment, pooled investigative site, and duration of predominant negative symptoms as fixed effects and baseline as covariate.
ASEN indicates asenapine; NA, not assessed; OLAN, olanzapine.
There were no significant between-group differences on any of the secondary efficacy measures at any time point in either of the core studies, including alternative negative symptom measures (ie, PANSS negative subscale and PANSS Marder negative factor scores), QLS or PANSS total score, or CGI-S (Table 1). Exploratory analysis revealed a significant correlation between improvements in NSA-16 and QLS total scores (EH: asenapine, r = -0.78; olanzapine, r = -0.75; WH: asenapine, r = -0.57; olanzapine, r = -0.39; all P < 0.0001; the inverse correlations reflect that improvement on the NSA-16 results in lower scores, whereas improvement on the QLS results in higher scores).
Although changes in positive symptoms with both treatments were small, PANSS positive subscale score changes were significantly greater with olanzapine than asenapine at weeks 12 through 26 in the EH and at weeks 20 and 26 in the WH (all P < 0.05; Fig. 1, C and D; Table 1). The PANSS general psychopathology subscale changes were greater with olanzapine than asenapine at week 20 in the EH (-5.8 ± 0.4 vs -4.5 ± 0.4; P < 0.05) and at week 26 in the WH (-4.5 ± 0.5 vs -3.0 ± 0.5; P = 0.02). In the EH core study, changes favored olanzapine over asenapine for the PANSS Marder hostility/excitement factor at weeks 20 (-0.3 ± 0.1 vs 0.1 ± 0.1; P < 0.05) and 26 (-0.3 ± 0.1 vs 0.2 ± 0.1; P < 0.05) and in the WH core study for the PANSS Marder positive symptom factor at week 26 (-2.0 ± 0.3 vs -0.8 ± 0.3; P = 0.004), disorganized thought factor at week 26 (-2.2 ± 0.3 vs -1.4 ± 0.3; P < 0.05), hostility/excitement factor at weeks 20 (-0.1 ± 0.2 vs 0.4 ± 0.2; P = 0.004) and 26 (-0.1 ± 0.2 vs 0.4 ± 0.2; P = 0.024), and anxiety/depression factor at weeks 20 (-0.9 ± 0.2 vs -0.3 ± 0.2; P = 0.014) and 26 (-1.1 ± 0.2 vs -0.3 ± 0.2; P = 0.001).
Week 26 changes in CGI-S score were similar for asenapine and olanzapine (EH: -0.8 ± 0.1 and -0.9 ± 0.1, respectively; WH: -0.5 ± 0.1 and -0.6 ± 0.1); similar proportions of subjects were classified as CGI-I responders at week 26. Based on a CMH test adjusted by PNS duration, response rates did not differ significantly between asenapine and olanzapine (EH: 45.9% and 54.9%; WH: 24.4% and 27.2%).
Overall depressive symptom changes with both treatments were minimal. However, olanzapine produced significantly greater CDSS total score improvements at weeks 4 through 26 compared with asenapine in the EH (all P < 0.05). Changes in CDSS scores did not differ at any time point in the WH study (Fig. 1, E and F).
Extension Studies
In the EH study, there was no significant group difference in change in NSA-16 total score at week 52 (asenapine, -16.9 ± 0.98; olanzapine, -15.4 ± 0.85; P = 0.23; Supplementary Table C, Supplemental Digital Content 4). In the WH study, the change from baseline in NSA-16 total score was significantly greater with asenapine compared with olanzapine at week 52 (-15.8 ± 1.48 vs -11.0 ± 1.27; P = 0.03; Supplementary Table C, Supplemental Digital Content 4). Significant differences were also observed in favor of asenapine at weeks 8, 16, and 42. This differential improvement was largely driven by those subjects whose duration of PNS was 2 years or less. In the EH study, similar results were observed with LOCF and OC analyses. In the WH study, the LOCF, but not the OC, analysis also demonstrated superiority of asenapine for negative symptoms.
In both extension studies, there were no significant between-group differences on the PANSS negative subscale, PANSS total, QLS, or CGI-S scores; in the WH extension study, significant differences in favor of asenapine were observed on the PANSS Marder negative symptom factor at weeks 42 and 52 using MMRM analysis (both P < 0.05; Supplementary Table C, Supplemental Digital Content 4). As in the core studies, significant correlations were observed between improvements in NSA-16 and QLS total scores for asenapine and olanzapine (EH: asenapine, r = -0.79; olanzapine, r = -0.77; WH: asenapine, r = -0.47; olanzapine, r = -0.61; all P < 0.0001).
Changes in positive symptoms were minimal. In the EH, LOCF analysis showed no significant between-group differences on the PANSS positive subscale score (Supplementary Table C, Supplemental Digital Content 4) or on the PANSS Marder hostility/excitement factor; OC analyses showed small but statistically significant between-group differences favoring olanzapine on change from core-study baseline to week 52 on the PANSS Marder hostility/excitement factor (asenapine: 0.0 ± 0.2; olanzapine: -0.5 ± 0.2; P < 0.05). In the Wh, PANSS positive subscale score changes were significantly greater with olanzapine at week 52 using LOCF analysis (asenapine, -0.4 ± 0.47; olanzapine, -1.5 ± 0.41; P = 0.04; Supplementary Table C, Supplemental Digital Content 4). There were no significant between-group differences in change from core-study baseline on the PANSS Marder factors for anxiety/depression, hostility/ excitement, or disorganized thought.
There were no significant asenapine/olanzapine differences in CGI-S change scores (EH: asenapine: -1.1± 0.09 and olanzapine: -1.1 ± 0.08; WH: asenapine: -0.9 ± 0.09 and olanzapine: -0.7 ± 0.08). Week 52 CGI-I response rates did not differ in the EH (asenapine, 57.0%; olanzapine, 61.0%; P = 0.69) or the WH (asenapine, 48.2%; olanzapine, 39.1%; P = 0.20).
Changes in depressive symptoms were minimal. On CDSS total score, there were no significant between-group differences in the EH or WH extension studies.
Safety and Tolerability
Core Studies
In the EH and WH, there were no substantive between-group differences in incidence of treatment-emergent or treatment-related AEs (Table 2). Most AEs were rated as mild or moderate. The incidence of treatment-related AEs was comparable across agents and studies (Table 2). Among those AEs reported at a rate of 5% or greater in the EH, the incidence of agitation and any EPS-related AE was greater than or equal to 2-fold higher with asenapine compared with olanzapine, and the incidence of increased weight was greater than or equal to 2-fold higher with olanzapine compared with asenapine (Table 2). In the WH, for those AEs reported at a rate of 5% or greater, the incidence of increased weight and dry mouth was greater than or equal to 2-fold higher with olanzapine compared with asenapine (see following paragraphs and Table 2).
TABLE 2.
Adverse Events Reported During the 6-Month Core Studies, Treated Subjects
| EH | WH | |||
|
Asenapine (n = 241) |
Olanzapine (n = 240) |
Asenapine (n = 244) |
Olanzapine (n = 224) | |
| Treatment-emergent AEs | 180 (74.7) | 165 (68.8) | 190 (77.9) | 184 (82.1) |
| Treatment-emergent SAEs | 26 (10.8) | 14 (5.8) | 28 (11.5) | 15 (6.7) |
| Treatment-related AEs | 133 (55.2) | 131 (54.6) | 158 (64.8) | 137 (61.2) |
| Treatment-related SAEs | 11 (4.6) | 8 (3.3) | 9 (3.7) | 7 (3.1) |
| DC due to treatment-emergent AEs* | 36 (14.9) | 17 (7.1) | 40 (16.4) | 30 (13.4) |
| DC due to treatment-related AEs | 30 (12.4) | 15 (6.3) | 30 (12.3) | 20 (8.9) |
|
Treatment-emergent AEs reported by ?5% of subjects† | ||||
| Insomnia | 38 (15.8) | 26 (10.8) | 43 (17.6) | 26 (11.6) |
| Headache | 31 (12.9) | 23 (9.6) | 33 (13.5) | 23 (10.3) |
| Somnolence | 30 (12.4) | 27 (11.3) | 36 (14.8) | 43 (19.2) |
| Anxiety | 23 (9.5) | 20 (8.3) | 26 (10.7) | 16 (7.1) |
| Dizziness | 9 (3.7) | 5 (2.1) | 18 (7.4) | 21 (9.4) |
| Sedation | 7 (2.9) | 9 (3.8) | 16 (6.6) | 17 (7.6) |
| Worsening of schizophrenia | 17 (7.1) | 9 (3.8) | 15 (6.1) | 12 (5.4) |
| Agitation | 15 (6.2) | 3 (1.3) | 10 (4.1) | 6 (2.7) |
| Nausea | 13 (5.4) | 9 (3.8) | 20 (8.2) | 11 (4.9) |
| Fatigue | 11 (4.6) | 16 (6.7) | 12 (4.9) | 8 (3.6) |
| Increased weight | 11 (4.6) | 51 (21.3) | 23 (9.4) | 48 (21.4) |
| Dry mouth | 4 (1.7) | 3 (1.3) | 9 (3.7) | 18 (8.0) |
| Increased appetite | 3 (1.2) | 6 (2.5) | 8 (3.3) | 12 (5.4) |
| EPS reported as AEs | ||||
| Any | 20 (8.3) | 8 (3.3) | 40 (16.4) | 27 (12.1) |
| Akathisia | 7 (2.9) | 3 (1.3) | 22 (9.0) | 13 (5.8) |
| Parkinsonism | 5 (2.1) | 4 (1.7) | 12 (4.9) | 10 (4.5) |
| Dyskinesia | 2 (0.8) | 1 (0.4) | 5 (2.0) | 2 (0.9) |
| Dystonia | 4 (1.7) | 3 (1.3) | 4 (1.6) | 1 (0.4) |
| Oculogyric crisis | 1 (0.4) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Bradykinesia | 2 (0.8) | 0 (0.0) | 1 (0.4) | 0 (0.0) |
| Gait disturbance | 1 (0.4) | 0 (0.0) | 1 (0.4) | 1 (0.4) |
| Tardive dyskinesia | 0 (0.0) | 0 (0.0) | 1 (0.4) | 2 (0.9) |
| Cogwheel rigidity | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (0.4) |
| Head titubation | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (0.4) |
*Discontinuations due to AEs based on AE case report forms; compare to Figure 1, showing discontinuations due to AEs based on end-of-treatment disposition case report forms.
† Includes SAEs.
DC indicates discontinuation.
Three deaths were reported in the EH study. One asenapine-treated subject committed suicide during the initial cross-titration period. A second asenapine subject was hospitalized with suspected tuberculosis 3 days after starting treatment and died of metastatic lung cancer. One olanzapine-treated subject committed suicide during the 30-day follow-up period. There were no deaths reported in the WH study.
The overall incidence of EPS-related AEs was higher in the WH than in the EH (Table 2). In the EH, the incidence of EPS-related AEs was higher with asenapine (8.3%; 95% confidence interval [CI], 5.1%-12.5%) than olanzapine (3.3%; 95% CI, 1.4%-6.4%), with an odds ratio of 2.6 (95% Wald CI, 1.1 -6.1); EPS-related AEs were also higher with asenapine (16.4%; 95% CI, 11.9%-21.6%) than olanzapine (12.1%; 95% CI, 8.1%-17.0%) in the WH, with an odds ratio of 1.4 (95% Wald CI, 0.9-2.4; Table 2). There were no discontinuations due to EPS-related AEs in the EH and 5 in the WH (asenapine, 3; olanzapine, 2). Akathisia and parkinsonism were the most common EPS-related AEs with asenapine and olanzapine. Among those EPS-related AEs reported at a rate of 2% or greater in either core study, the only difference of greater than 2-fold between treatments occurred with dyskinesia in the WH study (asenapine, 2.0%; olanzapine, 0.9%). There were no significant between-group differences in baseline ESRS-A total scores. In the EH study, there were no significant between-group differences in ESRS-A subscale changes; the only significant difference in the WH study was on the akathisia subscale score, in favor of olanzapine. Overall, changes from baseline were very small (± 0.2) for both asenapine and olanzapine on all ESRS-A total scales and subscales with the exception of the parkinsonism total score in the WH, in which changes from baseline ranged from -0.3 to -1.3 with asenapine and -0.1 to -1.7 with olanzapine.
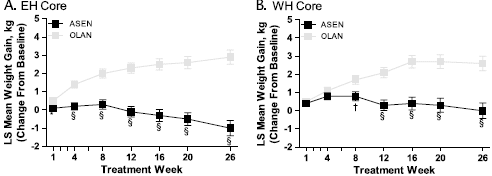
In the EH study, the incidence of clinically relevant weight gain (?7% increase from baseline to end point) was 7.9% with asenapine versus 24.6% with olanzapine; incidence of clinically relevant weight loss (?7% decrease from baseline to end point) was 11.6% with asenapine versus 3.3% with olanzapine. The between-group differences in weight gain were statistically significant from week 1 onward, with weight change (least squares [LS] mean ± SE) at week 26 being -1.0 ± 0.4 kg with asenapine and 2.9 ± 0.4 kg with olanzapine (P < 0.0001; Fig. 2A). In the WH study, the incidence of clinically relevant weight gain was 8.0% with asenapine versus 18.6% with olanzapine, and clinically relevant weight loss was 4.2% versus 3.2%. Between-group differences in weight gain were statistically significant from week 8 onward (week 26: 0.0 ± 0.4 kg with asenapine vs 2.6 ± 0.4 kg with olanzapine; P < 0.0001; Fig. 2B).
FIGURE 2. LS mean ± SE changes in body weight (MMRM analysis) in the 26-week core studies (A and B in the EH and WH regions, respectively) studies. ASEN indicates asenapine; OLAN, olanzapine. Based on an MMRM analysis, the model includes terms for treatment pooled investigative site, visit, treatment by visit and baseline, and duration of persistent negative symptom as covariates, with an unstructured variance-covariance matrix structure.
In both studies, most subjects remained in their baseline BMI category (<18.5, 18.5-24.99, 25.0-29.99, 30.0-34.99, 35.0-35.99, and ?40 kg/m2). Subjects treated with asenapine were less likely to shift to a higher BMI category and more likely to shift to a lower category than those treated with olanzapine. These findings are consistent with changes from baseline in waist circumference, which were lower with asenapine than with olanzapine (Supplementary Table D, Supplemental Digital Content 5).
In the EH study, the percentage of asenapine-treated subjects meeting the NCEP criteria for metabolic syndrome was 12.4% at baseline and was unchanged at end point (WH: 22.1% at baseline to 23.6% at end point), but rose from 10.4% at baseline to 14.4% at end point with olanzapine (WH: 20.5%-23.3%). In the EH study, among subjects who did not meet NCEP criteria at baseline, the prevalence of new-onset metabolic syndrome at end point was as follows: asenapine 7.1% versus olanzapine 9.7% (WH: asenapine, 8.9%; olanzapine 11.9%). There were no marked laboratory test abnormalities with either treatment (Supplementary Table D, Supplemental Digital Content 5).
Extension Studies
In the EH study, incidence rates of treatment-emergent and treatment-related AEs were 85.1% and 58.2% with asenapine (WH: 82.4% and 71.8%) and 74.4% and 58.1% with olanzapine (WH: 90.9% and 73.6%). Most AEs were rated as mild or moderate. In the EH, rates of treatment-emergent and treatment-related SAEs were 11.2% and 5.2% with asenapine (WH: 5.9% and 2.4%), and 4.1% and 0.6% with olanzapine (WH: 4.5% and 2.7%). No deaths were reported with either treatment in either extension study.
The incidence of specific AEs showed no notable change from the 6-month results; insomnia and somnolence were among the most commonly reported AEs. In the EH study, of those AEs reported at a rate of 5% or greater, greater than or equal to 2-fold higher rates of schizophrenia (asenapine, 9.7%; olanzapine, 4.7%), agitation (6.0%; 0.6%), depressive symptoms (6.0%; 2.3%), and increased blood creatinine phosphokinase (5.2%, 2.3) were reported for asenapine, and greater than or equal to 2-fold higher incidences of increased weight (olanzapine, 27.9%; asenapine, 6.0%), increased blood glucose (5.2%; 2.2%), and back pain (5.2%; 0.7%) were reported for olanzapine. In the WH study, among those AEs reported at a rate of 5% or greater, asenapine was associated with greater than or equal to 2-fold higher rates of diarrhea (asenapine, 8.2%; olanzapine, 3.6%) and irritability (7.1%; 0.9%), and olanzapine was associated with greater than or equal to 2-fold higher rates of headache (olanzapine, 14.5%; asenapine, 9.4%), sedation (10.0%; 4.7%), increased appetite (7.3%; 3.5%), and influenza (5.5%; 1.2%).
Extrapyramidal symptom-related AEs were described mainly as mild or moderate and were not responsible for any discontinuations. The overall incidence of EPS-related AEs was higher with asenapine in both studies (EH: 10.4% vs 4.1% with olanzapine; WH: 23.5% vs 10.0% with olanzapine). In the EH study, akathisia was twice as common with asenapine than with olanzapine (4.5% vs 1.2%); in the WH study, dyskinesia (4.7% vs 1.8%), dystonia (3.5% vs 0%), and tardive dyskinesia (2.4% vs 0.9%) were twice as common with asenapine. In the EH, there were no significant between-group differences in changes from core-study baseline to week 52 in the ESRS-A total score or subscale scores. There were no significant between-group differences in ESRS-A ratings at week 52 in the WH, but olanzapine showed an advantage over asenapine on the dystonia subscale score at weeks 30, 34, and 42 and end point (all P < 0.05).
In the EH study, the incidence of clinically significant weight gain with asenapine and olanzapine was 11.3% and 33.9%, respectively (clinically significant weight loss: 21.8% and 4.7%). Between-group differences in LS mean ± SE weight gain in the EH study were statistically significant from week 4 onward (all P < 0.01), with the change at week 52 (LS mean ± SE) being -1.4 ± 0.6 kg with asenapine and 4.0 ± 0.5 kg with olanzapine (P < 0.0001). In the WH study, the incidence of clinically significant weight gain was 17.6% with asenapine and 30.0% with olanzapine (clinically significant weight loss: 9.4% and 7.3%). The weight change difference was statistically less with asenapine at several time points (weeks 12-34 and 42; all P < 0.05), but not at week 52 (LS mean ± SE; asenapine, 1.2 ± 0.8 kg; olanzapine, 2.2 ± 0.7 kg; P = 0.35).
As in the core studies, most subjects in both extension studies remained in the same baseline BMI category, but olanzapine-treated subjects were more likely to shift to a higher BMI category and to show an increase in waist circumference (Supplementary Table D, Supplemental Digital Content 5). Most laboratory tests showed no marked abnormalities with either treatment (Supplementary Table D, Supplemental Digital Content 5). The incidence of hyperprolactinemia was generally higher with asenapine than with olanzapine. In the EH study, the percentage of subjects meeting NCEP criteria for metabolic syndrome shifted from 20.9% at baseline to 17.6% at end point with asenapine (WH: 23.5%-28.2%) and from 25.7% to 35.6% with olanzapine (WH: 20.0%-30.0%).Among subjects not meeting NCEP criteria at baseline, the incidence of new-onset metabolic syndrome at end point was 8.0% with asenapine versus 17.8% with olanzapine in the EH and 11.8% versus 14.5% in the WH.
Discussion
In contrast to our hypothesis, there were no significant differences between asenapine and olanzapine for the primary outcome measure (ie, superiority of asenapine over olanzapine in the change from baseline NSA-16 total score at week 26). Both drugs were associated with a significant reduction in PNS during the 26-week core studies. The fact that there were only minimal changes in positive symptoms, depressive symptoms, or EPS suggests the observed improvements in PNS were not due to changes in these causes of secondary negative symptoms. However, the lack of a placebo control precludes a definitive interpretation of the study results (see below). The improvement in negative symptoms was also significantly correlated with improved QLS scores; therefore, improvement in PNS may translate into improved quality of life and functionality.
The overall reduction of PNS with both treatments persisted throughout the extension studies. In fact, at week 52 of the WH extension, asenapine demonstrated superiority over olanzapine. However, these positive results need to be interpreted in view of the fact that only a relatively small subset of participants continued in the extension study. The results obtained through MMRM analysis were also observed with the LOCF and OC analyses.
Olanzapine was associated with significant improvement in both positive and depressive symptoms, although the magnitude of change was relatively small. Asenapine was associated with more treatment discontinuations than olanzapine, which may be related in part to the exclusion of potential participants who had previously been found to have an inadequate negative symptom response to olanzapine. The tolerability of both drugs was in line with previously described profiles.6,18,19 Insomnia, somnolence, and headache were among the most commonly reported AEs with asenapine; weight gain was the most commonly reported AE with olanzapine. Olanzapine treatment was associated with significantly greater weight gain, whereas EPS AEs were more frequently reported with asenapine.
Despite the length of the study, olanzapine treatment was not associated with significant changes in laboratory measures associated with increased cardiovascular and metabolic risk in people with schizophrenia previously reported to occur with olanzapine treatment.20,21 However, despite instructions from the investigators concerning food intake before testing, a large majority of blood samples for laboratory testing in the EH studies were nonfasting, which may have compromised our ability to detect changes in these variables.
A number of randomized, double-blind, clinical trials have explored the efficacy of second-generation antipsychotics for negative symptoms. However, few have used inclusion and exclusion criteria to select a study population with PNS, and few were designed to evaluate changes in negative symptoms in isolation from changes in other symptoms.3 For example, in 4 studies with amisulpride,22 25 the principal methodological issue was inadequate control of potential sources of secondary negative symptoms.
In previous studies involving individuals with PNS, olanzapine has shown inconsistent results: positive benefit in a clinical trial26 and 2 path analyses,19,27 but no benefit in another trial,28 and equivocal results compared with amisulpride.29 With respect to the efficacy of other classes of drugs in treating negative symptoms, glycine and D-cycloserine were found to be ineffective,30 but there is encouraging preliminary evidence for the selective monoamine oxidase B inhibitor selegiline.31,32
There are limitations to the design of the studies reported here that warrant discussion. Most importantly, a placebo group was not included in this antipsychotic monotherapy study because of ethical concerns associated with long-term discontinuation of active treatment in a clinically stable individual. Although the lack of a placebo arm makes it difficult to differentiate to what extent the observed reductions in PNS represent true treatment effects versus nonspecific effects secondary to participation in a clinical trial, it is unlikely that the reductions in PNS observed in these trials were due to spontaneous remission after discontinuation of a worse agent because PNS symptoms were stable for a 6-month period preceding the initiation of treatment with asenapine and olanzapine.
The other major potential concern relates to the use of the NSA-16 to assess the primary outcome. Although the reductions in NSA-16 total score in the core and extension studies were substantial, the NSA-16 has not been used in prior published studies. Therefore, it is difficult to compare the current results with those obtained with different negative symptom rating instruments in previous studies. However, the NSA-16 has been validated,14 and the other negative symptom assessment procedures used in the current study yielded the same pattern of results, which suggests that the results obtained with the NSA-16 were not idiosyncratic.
In summary, these studies comprise the first methodologically sound, large-scale, long-term, randomized clinical trials to evaluate the efficacy of second-generation antipsychotics for the treatment of PNS.3,33 The current studies showed that long-term treatment with asenapine produced improvement in PNS and quality of life that were similar in magnitude to those of olanzapine. Although this suggests that PNS may improve with pharmacological treatment, the primary objective of the core studies was not met because asenapine did not demonstrate superiority over olanzapine. The tolerability of both drugs was similar to previously published studies.6,18,19 Asenapine, in contrast to olanzapine, demonstrated a better tolerability profile with respect to weight gain but was more likely to cause EPS. Future studies, also using asenapine over a longer period, may provide more information about its full therapeutic potential for the negative symptoms of schizophrenia.
Acknowledgments
The authors thank Dr Milana Zivkov for her assistance in the development of this manuscript and Jacquelyn G. Wilson, PharmD, BCPP (Pfizer Inc), for her contributions during the conduct of these studies. Medical writing and editorial assistance were provided by Steve Tiger, PA, and Jane Phillips, PhD, of Complete Healthcare Communications, Inc (Chaddsford, Pa). This assistance was funded by Merck Sharp & Dohme Corp, a subsidiary of Merck & Co., Inc, Whitehouse Station, NJ.
Author disclosure information
Dr Buchanan has been an advisory board member for Abbott, Astellas, AstraZeneca, Merck, Pfizer, Roche, Schering-Plough (now Merck; nonpaid), Solvay Pharmaceuticals, Inc, Takeda, and Wyeth; has served as a consultant for Abbott, Cypress Bioscience, GlaxoSmithKline, Sanofi-Aventis, and Takeda; and has been a DSMB member for Pfizer, Cephalon, and Otsuka. Dr Schooler has received research support from AstraZeneca, Bristol-Meyers Squibb, Eli Lilly & Company, H. Lundbeck, OrthoMcNeil Janssen, and Pfizer; and has been a consultant/ advisory board member for Abbott, Dainippon Sumitomo, Eli Lilly & Company, Hoffman LaRoche, H. Lundbeck, Pfizer, Schering-Plough (now Merck), and OrthoMcNeil Janssen. Drs Cazorla, Zhao, Phiri, den Hollander, Ha, Kouassi, and Szegedi are full-time employees of Merck. Dr Panagides was an employee of Schering-Plough (now Merck) at the time of the study. Dr Alphs was employed at Pfizer Inc at the time this research was conducted and is owner of the copyright to the NSA-16.
REFERENCES
1. Kirkpatrick B, Buchanan RW, Ross DE, et al. A separate disease within the syndrome of schizophrenia. Arch Gen Psychiatry. 2001;58(2):165-171.
2. Milev P, Ho BC, Arndt S, et al. Predictive values of neurocognition and negative symptoms on functional outcome in schizophrenia: a longitudinal first-episode study with 7-year follow-up. Am J Psychiatry. 2005;162(3):495-506.
3. Buchanan RW. Persistent negative symptoms in schizophrenia: an overview. Schizophr Bull. 2007;33(4):1013-1022.
4. Kirkpatrick B, Fenton WS, Carpenter WT Jr, et al. The NIMH-MATRICS consensus statement on negative symptoms. Schizophr Bull. 2006;32(2):214-219.
5. Kane J. Commentary: consensus statement on negative symptoms. Schizophr Bull. 2006;32:223-224.
6. Potkin SG, Cohen M, Panagides J. Efficacy and tolerability of asenapine in acute schizophrenia: a placebo- and risperidone-controlled trial. J Clin Psychiatry. 2007;68(10):1492-1500.
7. Laughren T, Levin R. Food and Drug Administration perspective on negative symptoms in schizophrenia as a target for a drug treatment claim. Schizophr Bull. 2006;32(2):220-222.
8. Sheehan DV, Lecrubier Y, Sheehan KH, et al. The Mini-International Neuropsychiatric Interview (M.I.N.I.): the development and validation of a structured diagnostic psychiatric interview for DSM-IV and ICD-10. J Clin Psychiatry. 1998;59(suppl 20):22-33.
9. Kay SR, Fiszbein A, Opler LA. The Positive and Negative Syndrome Scale (PANSS) for schizophrenia. Schizophr Bull. 1987;13(2):261-276.
10. Marder SR, Davis JM, Chouinard G. The effects of risperidone on the five dimensions of schizophrenia derived by factor analysis: combined results of the North American trials. J Clin Psychiatry. 1997;58(12):538-546.
11. Chouinard G, Ross A, Annable L, et al. Extrapyramidal Symptom Rating Scale [ESRS] (1980). In: Sajatovic M, Ramirez L, eds. Rating Scales in Mental Health. Hudson, OH: Lexi-Comp; 2001:209-213.
12. Addington D, Addington J, Schissel B. A depression rating scale for schizophrenics. Schizophr Res. 1990;3(4):247-251.
13. Guy W. Clinical Global Impressions. In: Guy W, ed. ECDEU Assessment Manual for Psychopharmacology. Washington, DC: US Department of Health, Education, and Welfare; 1976:217-222.
14. Alphs LD, Summerfelt A, Lann H, et al. The negative symptom assessment: a new instrument to assess negative symptoms of schizophrenia. Psychopharmacol Bull. 1989;25(2):159-163.
15. Heinrichs DW, Hanlon TE, Carpenter WT Jr. The Quality of Life Scale: an instrument for rating the schizophrenic deficit syndrome. Schizophr Bull. 1984;10(3):388-398.
16. Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. Executive summary of the third report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). JAMA. 2001;285(19):2486-2497.
17. Cui L, Hung HM, Wang SJ. Modification of sample size in group sequential clinical trials. Biometrics. 1999;55(3):853-857.
18. Schoemaker J, Naber D, Vrijland P, et al. Long-term assessment of asenapine vs. olanzapine in patients with schizophrenia or schizoaffective disorder. Pharmacopsychiatry. 2010;43(4):138-146.
19. Alvarez E, Ciudad A, Olivares JM, et al. A randomized, 1-year follow-up study of olanzapine and risperidone in the treatment of negative symptoms in outpatients with schizophrenia. J Clin Psychopharmacol. 2006;26(3):238-249.
20. Hennekens CH, Hennekens AR, Hollar D, et al. Schizophrenia and increased risks of cardiovascular disease. Am Heart J. 2005;150(6):1115-1121.
21. Newcomer JW. Antipsychotic medications: metabolic and cardiovascular risk. J Clin Psychiatry. 2007;68(suppl 4):8-13.
22. Paillere-Martinot ML, Lecrubier Y, Martinot JL, et al. Improvement ofsome schizophrenic deficit symptoms with low doses ofamisulpride. Am J Psychiatry. 1995;152(1):130-134.
23. Loo H, Poirier-Littre MF, Theron M, et al. Amisulpride versus placebo in the medium-term treatment of the negative symptoms of schizophrenia. Br J Psychiatry. 1997;170:18-22.
24. Danion JM, Rein W, Fleurot O. Improvement of schizophrenic patients with primary negative symptoms treated with amisulpride. Amisulpride Study Group. Am J Psychiatry. 1999;156(4):610-616.
25. Speller JC, Barnes TR, Curson DA, et al. One-year, low-dose neuroleptic study of in-patients with chronic schizophrenia characterised by persistent negative symptoms. Amisulpride v. haloperidol. Br J Psychiatry. 1997;171:564-568.
26. Lindenmayer JP, Khan A, Iskander A, et al. A randomized controlled trial of olanzapine versus haloperidol in the treatment of primary negative symptoms and neurocognitive deficits in schizophrenia. J Clin Psychiatry. 2007;68:368-379.
27. Tollefson GD, Sanger TM. Negative symptoms: a path analytic approach to a double-blind, placebo- and haloperidol-controlled clinical trial with olanzapine. Am JPsychiatry. 1997;154(4):466Y474.
28. Kopelowicz A, Zarate R, Tripodis K, et al. Differential efficacy of olanzapine for deficit and nondeficit negative symptoms in schizophrenia. Am JPsychiatry. 2000;157(6):987Y993.
29. Lecrubier Y, Quintin P, Bouhassira M, et al. The treatment of negative symptoms and deficit states of chronic schizophrenia: olanzapine compared to amisulpride and placebo in a 6-month double-blind controlled clinical trial. Acta Psychiatr Scand. 2006;114:319-327.
30. Buchanan RW, Javitt DC, Marder SR, et al. The Cognitive and Negative Symptoms in Schizophrenia Trial (CONSIST): the efficacy of glutamatergic agents for negative symptoms and cognitive impairments. Am J Psychiatry. 2007;164(10):1593-1602.
31. Amiri A, Noorbala AA, Nejatisafa AA, et al. Efficacy of selegiline add on therapy to risperidone in the treatment of the negative symptoms of schizophrenia: a double-blind randomized placebo-controlled study. Hum Psychopharmacol. 2008;23(2):79Y86.
32. Bodkin JA, Siris SG, Bermanzohn PC, et al. Double-blind, placebo-controlled, multicenter trial of selegiline augmentation of antipsychotic medication to treat negative symptoms in outpatients with schizophrenia. Am JPsychiatry. 2005;162(2):388Y390.
33. Alphs L, Panagides J, Lancaster S. Asenapine in the treatment of negative symptoms of schizophrenia: clinical trial design and rationale. Psychopharmacol Bull. 2007;40(2):41Y53.



