Клинико-психологический подход к проблеме митохондриальной патологии
Статьи
![]()
ЖУРНАЛ "ПРАКТИКА ПЕДИАТРА"
Опубликовано в журнале: «ПРАКТИКА ПЕДИАТРА»; март-аперль; 2015; стр. 50-56.
А.А. Беликова, зав. отделением мед. психологии - мед. психолог в ГКБ № 1 им. Н.И. Пирогова, Московский городской психолого-педагогический университет
В настоящее время изучение митохондриальной патологии вызывает неподдельный интерес медицинского и психологического сообществ. За последние десятилетия появились работы, посвященные процессу нарушения клеточного энергообмена, сосредоточенного в органеллах синтеза АТФ - митохондриях. К настоящему времени разработкой проблем митохондриальной патологии с клинических позиций активно занимаются Д.А. Харламов, Е.А. Николаева, В.С. Сухоруков и И.В. Леонтьева в Научно-исследовательском клиническом институте педиатрии, Т.С. Угольник, И.В. Манаенкова в Гомельском государственном медицинском университете, Н.С. Прохорова, Л.А. Демиденко в Крымском государственном медицинском университете и др.
Ключевые слова: митохондриальные болезни, митохондриальная ДНК и ядерная ДНК, миопатии, энцефаломиопатии, расстройства психологического развития.
Key words: mitochondrial diseases, mitochondrial DNA and nuclear DNA, myopaties, encephalomyopathies, psychological developmental disorders.
Нарушения функционирования митохондрий приходятся на ранние периоды повреждения клеток. Понятие «митохондриальная патология» сформировалось в медицине к концу ХХ века благодаря выявленным в клинической генетике мутациям генов, ответственным за синтез митохондриальных белков. В первую очередь были изучены болезни, связанные с мутациями митохондриальной ДНК, открытой в 1960-х годах. Однако дефекты митохондрий могут быть связаны и с мутациями ядерной ДНК (нарушение ядерно-митохондриальных взаимодействий). По данным зарубежных исследователей, частота этих заболеваний у новорожденных составляет 1:5000.
Нарушение клеточной энергетики в так называемых дыхательных станциях приводит к полисистемным заболеваниям, клинические проявления которых весьма разнообразны.
В настоящее время можно выделить две группы заболеваний, возникновение которых обусловлено мутациями митохондриальной ДНК и ядерной ДНК соответственно.
К первой группе можно отнести синдром Кернса - Сейра, синдром Пирсона синдром MELAS, синдром MERRF, синдром NAPR, синдром Лебера - оптическая нейропатия и синдром множественных делеций.
Ко второй группе - атаксию Фридриха, синдром Вольфрама, синдром Лея, синдром Альперса, митохондриальные заболевания, обусловленные нарушением цикла Кребса, митохондриальные болезни, обусловленные нарушением бета-окисления жирных кислот с различной длиной углеродной цепи, и синдром Люфта (Luft disease) - гиперметаболизм нетиреоидного происхождения.
Однако единой классификации митохондриальных болезней на сегодняшний момент нет. Условно заболевания можно делить на «первичные» и «вторичные».
«Первичные» связаны с ярко выраженными наследственными синдромами, обусловленными мутациями генов, ответственных за митохондриальные белки. «Вторичные» митохондриальные заболевания включают в себя нарушение клеточного энергообмена как важное звено формирования патогенеза.
Становление науки митохондриологии произвело революцию в современных представлениях о медицинских аспектах энергетического обмена у человека.
Одним из главных достижений медицинской митохондриологии явилось создание эффективного диагностического инструментария (клинические, биохимические, морфологические и молекулярно-генетические критерии митохондриальной недостаточности), позволившего оценивать полисистемные нарушения клеточного обмена и в дальнейшем подбирать индивидуальную эффективную симптоматическую лечебно-медикаментозную терапию.
Клинический полиморфизм, сложность диагностики и тяжесть течения определяют актуальность митохондриальных болезней для врачей различных специальностей.
Однако с психологической точки зрения данные заболевания не изучались, в частности в коррекционной и клинической психологии неизвестно каких-либо определенных исследований по данной проблеме. В связи с этим огромное количество вопросов на сегодняшний момент являются открытыми и, безусловно, представляют интерес.
Интересные факты о митохондриальной патологии
Первые митохондриальные заболевания были описаны раньше, чем открыта ДНК в митохондриях. В 1958 году был открыт синдром Кернса - Сейра, в 1962 году - болезнь Люфта: нетироидальный гиперметаболизм (всего 2 случая за 40 лет). В 1981 году был расшифрован митохондриальный геном человека (Anderson et al.). А в 1988 году были идентифицированы первые патогенные мутации мтДНК (Holt et al., Wallace et al.).
Интересно что, в ядре около 70 000 генов в двух копиях каждый. Гены составляют менее 1% от всей ядерной ДНК. А в митохондриях - 37 генов в тысячах копий каждый. Гены составляют более 92% от всей митохондриальной ДНК.
Мутации в митохондриальной ДНК человека происходят в 5 раз чаще, чем в ядерной. В настоящее время описано более 190 патогенных точечных мутаций митохондриальной ДНК и около 200 делеций, инсерций и других структурных реорганизаций мтДНК. Некоторые мутации ядерной ДНК могут приводить к мутациям митохондриальной ДНК: ген ДНК-полимеразы-гамма (осуществляет синтез мтДНК); ген тимидинфосфорилазы (нарушает метаболизм тимидина); ген Twinkle (участвует в поддержании целостности митохондриального генома). Информация о мутациях в этих генах накапливается стремительно.
Ткани с низким порогом мутантной ДНК:
Клетки этих тканей наиболее метаболически активны, энергетически зависимы.
Поскольку самые энергоемкие это нервные и мышечные клетки, при МБ наиболее распространены мышечные и неврологические проблемы, такие как мышечная слабость, непереносимость физических нагрузок, потеря слуха, нарушения баланса и координации, эпиприступы и проблемы с обучением. Другие частые осложнения - нарушения зрения, дефекты сердца, диабет и задержка роста. Обычно у ребенка с МБ присутствуют два или более из этих симптомов, некоторые из них настолько часто встречаются вместе, что сгруппированы в синдромы МБ, вызывающие выраженные мышечные проблемы. Эти синдромы именуют митохондриальными миопатиями (myo означает «мышца», а pathos -«болезнь»), а те, которые вызывают как мышечные, так и неврологические проблемы, -митохондриальными энцефаломиопатиями (encephalo - «мозг»).
Несмотря на многочисленные потенциальные проблемы, МБ не всегда приводят к серьезной инвалидизации. Иногда здоровых митохондрий бывает достаточно для компенсации действия поврежденных. Кроме того, поскольку некоторые симптомы МБ (такие как диабет и сердечная аритмия) часто встречаются в общей популяции, для них имеется эффективная медикаментозная терапия (инсулин или противоаритмические препараты).
Общая характеристика митохондриальной патологии
Митохондриальные болезни (МБ) являются результатом нарушения функционирования системы окислительного фосфорилирования, которая состоит из четырех ферментных комплексов, (комплексы I-IV), расположенных во внутренней мембране митохондрий и транспортирующих электроны по дыхательной цепи. Пятый комплекс (комплекс V) представляет собой фермент АТФ-синтазу, которая, используя протонный градиент, производит АТФ. Повреждение этого процесса может поразить каждый орган и каждую систему. Эти полиорганность и полисистемность делают диагностику и классификацию заболеваний дыхательной цепи особенно затруднительной. У пациентов могут наблюдаться разнообразные симптомы, которые часто не вписываются в рамки определенных клинических фенотипов.
Митохондриальные болезни могут быть спорадическими или передаваться разными типами наследования: материнским, аутосомно-доминантным, аутосомно-рецессивным и Х-сцепленным.
Очевидно, что при энергетических нарушениях наиболее страдают ткани с высокой энергетической потребностью, что и происходит при митохондриальных заболеваниях, при которых повреждение мышц и периферических нервов наблюдается достаточно часто.
Происхождение митохондриальных болезней
Прежде всего, МБ - не инфекционные заболевания и не являются следствием внешнего воздействия. Они обусловлены мутациями, или «поломками» в генах - клеточных матрицах для производства протеинов.
Гены ответственны за формирование и развитие человеческого организма и передаются от родителей к детям вместе с присутствующими в них мутациями или дефектами. Это значит, что МБ - наследственные состояния, хотя могут проявляться у членов одной семьи по-разному.
Дефицит одного или нескольких митохондриальных комплексов может быть причиной МБ. Например, наследственная оптическая невропатия Лебера, при которой описаны три мутации комплекса I. У большинства пациентов патологические изменения ограничиваются зрительным нервом, в частности отсутствуют изменения в мышечной ткани.
Когда клетка заполнена дефектными митохондриями, она не только лишена АТФ, но и в ней могут накапливаться неиспользуемые молекулы топлива и кислород, что приводит к катастрофическим последствиям.
В этом случае избыточные молекулы топлива используются для синтеза АТФ неэффективно, в результате чего могут образовываться потенциально опасные продукты, такие как молочная кислота (это также происходит, когда клетки испытывают недостаток кислорода, например мышечные клетки при усиленных физических нагрузках). Накопление молочной кислоты в крови - лактатацидоз - может вызывать повреждение нервной и мышечной тканей.
При этом неиспользуемый в клетке кислород может трансформироваться в разрушительные соединения, именуемые реактивными формами кислорода, включая т. н. свободные радикалы (они являются мишенью для т. н. антиоксидантных препаратов и витаминов).
Синтезированная в митохондриях АТФ -основной источник энергии для сокращения мышечных и возбуждения нервных клеток. Таким образом, нервные и мышечные клетки особенно чувствительны к дефектам митохондрий. Сочетание дефицита энергии и накопления токсинов в этих клетках играет одну из основных ролей в развитии симптомов митохондриальных миопатий и энцефаломиопатий.
Синдромы митохондриальной патологии
Обычно эти синдромы наследуются или по типу материнского наследования, или по т. н. менделевскому типу, а также могут быть спорадическими, что означает, что они возникают при отсутствии семейной истории.
Синдром Кернса - Сейра (КСС, KSS)
Тип наследования: спорадический. Возраст дебюта: до 20 лет.
Особенности: Это заболевание определено прогрессирующей наружной офтальмоплегией (обычно в качестве начального симптома) и пигментарной ретинопатией, пигментацией сетчатки типа «соль с перцем», что влияет на зрение, но нередко и не оказывает на него влияния. Другие частые симптомы - блок проводимости (в сердце) и атаксия. Менее типичные симптомы - умственная отсталость или задержка умственного развития, замедленное половое созревание и низкий рост.
Синдром Лея: подострая некротизирующая энцефаломиопатия (СЛМН - синдром Лея с наследованием по материнской линии, MILS)
Тип наследования: материнский, менделевский. Возраст дебюта: младенчество.
Особенности: Синдром Лея вызывает аномалии головного мозга, что может привести к атаксии, эпиприступам, ослабленному зрению и слуху, задержкам развития и нарушению контроля дыхания. Синдром также вызывает мышечную слабость с наибольшим эффектом на глотание, речь и движения глаз.
Синдром делеции митохондриальной ДНК (СДМ, MDS)
Тип наследования: менделевский. Возраст дебюта: младенчество.
Особенности: Это заболевание обычно вызывает мышечную слабость и/или патологию печени и намного реже - мозговые аномалии. Мышечная вялость, трудности с питанием и задержки развития - наиболее частые симптомы; реже - прогрессирующая наружная офтальмоплегия и эпиприступы. Может быть кардиомиопатия с судорогами (синдром Де Тони - Дебре - Фанкони).
Митохондриальная энцефаломиопатия, лактатацидоз, инсультоподобные эпизоды (МЭЛАС, MELAS)
Тип наследования: материнский. Возраст дебюта: детство - подростковый возраст.
Особенности: МЭЛАС вызывает периодические инсультоподобные эпизоды в головном мозге, мигренеподобные головные боли, рвоту, эпиприступы и может привести к необратимым поражениям головного мозга. К другим частым симптомам относятся прогрессирующая наружная офтальмоплегия, генерализованная мышечная слабость, непереносимость физических нагрузок, потеря слуха, диабет и низкорослость.
Миоклонус-эпилепсия с «рваными красными волокнами» (МЭРРФ, MERRF)
Тип наследования: материнский. Возраст дебюта: позднее детство - юность.
Особенности: Наиболее выраженные симптомы - миоклонус (подергивания мышц), эпиприступы, атаксия и мышечная слабость. Могут также наблюдаться ухудшение слуха и низкорослость.
Митохондриальная нейрогастроинтестинальная энцефаломиопатия (МНГИЭ, MNGIE)
Тип наследования: менделевский. Возраст дебюта: обычно до 20 лет.
Особенности: Это заболевание вызывает ПНО, птоз, слабость конечностей и гастроинтестинальные (пищеварительные) проблемы, включая хроническую диарею и абдоминальные боли. Другой частый симптом - периферическая нейропатия (нарушение функционирования нервов, которое может привести к ухудшению чувствительности и мышечной слабости).
Нейропатия, атаксия и пигментозный ретинит (НАПР, NARP)
Тип наследования: материнский. Возраст дебюта: младенчество - подростковый возраст.
Особенности: НАПР вызывает нейропатию, атаксию и пигментозный ретинит (дегенерацию сетчатки глаза, что приводит к потере зрения). Наблюдаются также задержка развития, эпиприступы и деменция.
Синдром Пирсона
Тип наследования: спорадический. Возраст дебюта: младенчество.
Особенности: Этот синдром вызывает тяжелую анемию и нарушение функции поджелудочной железы. У выживших детей обычно впоследствии развивается синдром Кернса - Сейра.
Прогрессирующая наружная офтальмоплегия (ПНО, PEO)
Тип наследования: материнский, менделевский, спорадический. Возраст дебюта: обычно подростковый возраст - юность.
Особенности: Как отмечалось выше, ПНО -частый симптом митохондриальных заболеваний, однако иногда его выделяют в самостоятельный синдром. Часто ассоциирован с непереносимостью физических нагрузок.
Синдром Лебера: LHON (наследственная атрофия зрительных нервов - 1871 г.)
Тип наследования: материнский. Возраст дебюта: 20-30 лет.
Особенности: потеря зрения происходит у людей вследствие атрофии зрительного нерва и дегенерации ганглиозного слоя клеток сетчатки. Заболевание связано с передаваемой от матери мутацией митохондриальной ДНК в одном из ND генов (комплекс I).
Загадки синдрома Лебера: В 80-85% случаев поражаются мужчины. Лишь у 50% мужчин и 10% женщин - носителей патогенных мутаций комплекса I в действительности происходит потеря зрения.
Чаще всего мутации, ведущие к синдрому Лебера, встречаются в мтДНК гаплогруппы J; эту группу несут около 15% европейцев.
Диагностика МБ
Ни один из отличительных симптомов митохондриального заболевания - мышечная слабость, непереносимость нагрузок, ухудшение слуха, атаксия, эпиприступы, неспособность к обучению, катаракта, дефекты сердца, диабет и низкорослость - не является уникальным именно для такого заболевания. Однако комбинация трех или более из этих симптомов у одного индивида свидетельствует в пользу МБ, особенно если симптомы затрагивают более одной системы организма.
Диагностические тесты при митохондриальных заболеваниях
Физикальное обследование обычно включает в себя тесты на силу и выносливость, такие, например, как повторяющиеся сжатия-разжатия кулака или подъем и спуск по небольшой лестнице. Неврологическое обследование может включать в себя проверку рефлексов, зрения, речи и базовых когнитивных (познавательных) способностей.
В зависимости от полученной на этом первом этапе информации, врач может назначить более специализированные пробы, способные выявить аномалии в мышцах, головном мозге и других органах.
Наиболее важный из таких тестов - мышечная биопсия, заключающаяся в извлечении небольшого образца мышечной ткани для исследования. В нормальных мышцах человека митохондрии локализуются под сарколеммой и в пространстве между миофибриллами, где они примыкают к I полоскам. Признаком митохондриальных нарушений в мышцах у большинства, но не у всех пациентов являются «рваные красные волокна» (ragged-red fibers, -RRF), которые выявляются при трихромной окраске по модифицированному методу Гомори. Однако окраска на сукцинатдегидрогеназу, которая проявляется окрашиванием темно-синего цвета, - более чувствительный и точный метод, выявляющий эти скопления митохондрий. Появление RRF является следствием пролиферации субсарколеммальных и межмиофибриллярных аномальных митохондрий. Другие красители могут выявить отсутствие в мышцах важных митохондриальных энзимов. Можно также выделить из мышц митохондриальные протеины и измерить их активность.
В дополнение к мышечной биопсии могут использоваться другие неинвазивные (не требующие извлечения образцов тканей) методы исследований. Например, с помощью метода, именуемого фосфорной магнитно-резонансной спектроскопией (МРС), можно измерить уровни фосфокреатина и АТФ (которые часто снижены в пораженных МБ мышцах).
Для неинвазивного тестирования используют также седимент мочи, соскоб внутренней поверхности щеки, реже - волосяные фолликулы.
Компьютерная и магнитно-резонансная томография (КТ и МРТ) могут быть проведены для визуального выявления признаков повреждений в головном мозге, а поверхностные электроды на коже головы могут использоваться для получения записи биоэлектрической активности головного мозга, именуемой электроэнцефалограммой.
Аналогичные методики могут быть использованы для проверки функционирования других органов и тканей организма. Например, электрокардиограмма (ЭКГ) позволяет проверить активность сердца, а анализ крови может выявить признаки дисфункции почек.
Наконец, генетический тест может определить, есть ли у пациента генетическая мутация, обусловливающая развитие митохондриального заболевания. Лучше всего для этого теста использовать генетический материал, выделенный из образца крови или мышечного биоптата. Важно осознавать, что, хотя положительный результат может подтвердить диагноз, отрицательный не обязательно его опровергает. В случае еще не описанных, редких «private» мутаций проводят прямое секвенирование мтДНК.
Основные проблемы, ассоциированные с МБ, - низкая энергия, выработка свободных радикалов и лактатацидоз - могут привести к развитию различных симптомов во многих различных органах. На рисунке показаны общие симптомы МБ, специфическая совокупность которых представлена у большинства людей с этими заболеваниями. Многие из этих симптомов поддаются лечению.
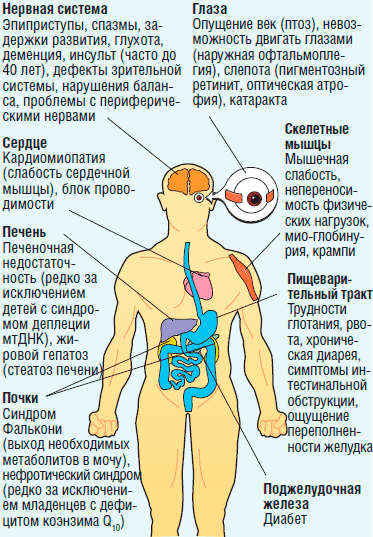
Рис. Митохондриальное заболевание: взгляд изнутри
Специфика когнитивных функций у детей с митохондриальной патологией
В диссертационном исследовании А.И. Крапивкина на соискание ученой степени доктора медицинских наук, выполненном в Москве в 2012 году, на тему «Патогенетическая характеристика важнейших клинических форм нарушений психологического развития у детей и оптимизация их лечения» отдельно анализировались расстройства психологического развития и поведения у детей с митохондриальными болезнями.
Группу детей с митохондриальными энцефаломиопатиями составил 21 ребенок, со следующими диагнозами: митохондриальная энцефаломиопатия (n=12), синдромом MERRF (n=1), синдромом Кернса - Сейра (n=4) и синдромом MELAS (n=4), проходивший стационарное обследование в Федеральном государственном бюджетном учреждении «Московский научно-исследовательский институт педиатрии и детской хирургии» Минздравсоцразвития Российской Федерации.
Анализ клинических проявлений нарушений тканевого обмена выявил высокую частоту поражения нервной и нервно-мышечной системы у обследованных детей. У всех отмечались низкая переносимость физической нагрузки, мышечная слабость, быстрая утомляемость.
Большинство детей имели выраженную неврологическую симптоматику, и только у 14% выявлены минимальные отклонения при неврологическом обследовании. Координаторные расстройства определялись у большинства больных и проявлялись в виде моторной неловкости, атактической походки, затруднений при выполнении координаторных проб. При электроэнцефалографическом исследовании у 2 пациентов отмечено «замедление» основной активности головного мозга, у остальных отмечены изменения дисфункционального характера. При нейрорадиологическом обследовании только у 4 детей были выявлены структурные изменения: у 2 детей - последствия инсультоподобных эпизодов, у 2 - проявления лейкомаляции.
Значительный интеллектуальный дефицит (IQ <70) был установлен у 35% детей. Расстройства психологического развития и поведения были выявлены у всех детей. Нарушения речевого развития имели 95%, из них 37% - умеренного характера. Нарушения психического развития установлены у 68,4% пациентов, из них у 38,5% - умеренного, а у 15% - выраженного характера. При проведении психологического тестирования у детей отмечались выраженные нарушения кратковременной и отсроченной памяти, сложности с концентрацией и переключением внимания, значительные нарушения зрительной памяти. Гетерогенность нарушений психологического развития и поведения при этом, очевидно, была связана с нозологической гетерогенностью детей с первичными митохондриальными заболеваниями.
Таким образом, выраженные нарушения психологического развития и поведения фактически всегда сопровождают клиническую картину митохондриальных заболеваний. В то же время, как правило, при данных болезнях неврологические расстройства привлекают основное внимание и выходят на первый план при клинической оценке нарушений.
Заключение
Изучение митохондриальных заболеваний в парадигмах психологической науки - новый этап в развитии митохондриологии, а также процесс обогащения и накопления опыта и знаний, собственно, психологией.
Нарушение энергообмена в молекулах синтеза АТФ приводит к весьма гетерогенным и полисистемным заболеваниям с разнородными клиническими проявлениями.
Специфика развития когнитивной сферы у детей с митохондриальной патологией характеризуется изменениями кратковременной и отсроченной вербальной и зрительной памяти, концентрации и переключении внимания, характер и степень выраженности которых зависят от нозологической формы расстройства психологического развития и поведения.
Исходя из вариабельности представленных нарушений, можно предположить, что недостаточность клеточного энергообмена более выражена у детей с митохондриальной патологией с нарушениями интеллекта и непосредственно влияет на когнитивные функции. В зависимости от зональности энергообмена когнитивные нарушения представлены в разной степени.
Оценка характера нарушений при данных заболеваниях позволит разработать коррекционно-реабилитационные методы, направленные на выявление ресурсного потенциала детей с митохондриальной патологией. Таким образом, это повысит социализацию детей в учреждениях общего и специального дошкольного и школьного образования, повысит качество жизни детей и сформирует в их семьях целостное представление о реабилитационных мероприятиях и психолого-педагогическом воздействии.



