Гипермобильный синдром: клинические проявления, дифференциальный диагноз, подходы к терапии
СтатьиН.Г. Правдюк, Н.А.Шостак
Кафедра факультетской терапии им. А.И. Нестерова, Российский государственный медицинский университет, Москва Дисплазия соединительной ткани (ДСТ) представляет собой уникальную онтогенетическую аномалию развития организма, которая относится к числу сложных вопросов современной медицины. Рассматриваются основные подходы к дифференциальной диагностике различныхформ ДТС. Ведущее место отводится клинической оценке, подходам к терапии одного из вариантов недифференцированной формы ДТС – гипермобильному синдрому.
Ключевые слова: дисплазия соединительной ткани, гипермобильный синдром, гипермобильность суставов.
РФК 2008;3:70-75
Hypermobility syndrome: clinical manifestations, differential diagnosis, therapy approaches
N.A. Shostak, N.G. Pravdyuk
Chair of Faculty Therapy named after A.I. Nesterov, Russian State Medical University, Moscow Connective tissue dysplasia (CTD) represents special ontogenetic abnormality which is a complex problem of contemporary medicine. The principles of differential diagnosis of various forms of CTD are considered. A clinical estimation and therapy approaches are discussed with focus on hypermobility syndrome as one of undifferentiated form of CTD.
Key words: connective tissue dysplasia, hypermobility syndrome, joint hypermobility.
Rational Pharmacother. Card. 2008;3:70-75
Группа наследственных заболеваний соединительной ткани и скелета была впервые выделена американским генетиком Mc Kusick в 1955 году. К тому времени она объединяла лишь некоторые нозологические формы: несовершенный остеогенез, синдром Марфана, синдром Элерса-Данло, эластическую псевдоксантому и гаргоилизм. [1] В течение последующих трех десятилетий благодаря достижениям генетики были описаны и классифицированы свыше 200 заболеваний соединительной ткани и скелета наследственного характера.
Клинические проявления дисплазий соединительной ткани
Дисплазия соединительной ткани (ДСТ) – генетически детерминированное нарушение развития соединительной ткани, приводящее к изменению ее структуры и функций и реализующееся в клиническом многообразии фенотипических признаков и органных проявлений. В настоящее время предложено подразделение ДСТ на дифференцированный и недифференцированный синдромы [2].
Дифференцированные дисплазии характеризуются установленным генным или биохимическим дефектом с определенным типом наследования и клинической картиной заболевания (синдром Марфана, синдром Элерса-Данлоса, несовершенный остеогенез, эластическая псевдоксантома и др.).
Недифференцированные дисплазии соединительной ткани диагностируются в случаях, когда наборфенотипических признаков не соответствует ни одному из дифференцированных синдромов.
Соединительнотканная дисплазия, затрагивая все органы и системы, проявляется комплексом фенотипических признаков. Весьма характерным для ДСТ является генерализованная гипермобильность суставов (ГМС), которая может быть ведущим признаком как недифференцированной ДСТ, так и частью дифференцированных синдромов.
Для объективной оценки генерализованной гипермобильности суставов используются критерии Бейтона [3] (табл. 1).
Таблица 1. Признаки гипермобильности суставов (критерии Бейтона)
|
Гипермобильность оценивают в баллах: 1 балл означает патологическое переразгибание в одном суставе на одной стороне. Максимальная величина показателя, учитывая двухстороннюю локализацию, - 9 баллов (8 - за 4 первых пункта и 1 - за 5-й пункт). Показатель от 4 до 9 баллов расценивается как состояние гипермобильности.
Научный и практический интерес к ГМС возник еще в конце 19 века, когда были описаны наследственные синдромы, в клинической картине которых ГМС являлась одним из ведущих симптомов (синдром Марфана, синдром Элерса-Данло, несовершенный остеогенез). Влияние ГМС на состояние здоровья имеет самые разнообразные аспекты. Первое описание ГМС было сделано в 1967 году Kirk, Ansell и Bywaters. [4]. Авторами был предложен термин "гипермобильный синдром" (ГС), отражающий феномен гипермобильности суставов, сочетающийся с дисфункцией опорно-двигательного аппарата (подвывихи, артралгии). Позже стало известно, что ГМС ассоциируется с внешними фенотипическими признаками ДСТ, сходными с маркерами дисплазии при дифференцированных синдромах, а «гипермобильный синдром» стал рассматриваться в рамках нозологической формы. Однако генетическая основа ГС до настоящего времени остается неизвестной.
Клинические проявления ГС многообразны и включают как суставные, так и внесуставные признаки, отраженные в критериях ГС. Диагностические критерии ГС представлены в табл. 2 и именуются Брайтоновскими критериями [5].
Малые критерии ГС были дополнены в ходе работ А.Г. Беленького [6] и включают пролапс митрального клапана, полую стопу, браходактилию, деформацию грудной клетки, сандалевидную щель стопы, сколиоз, Hallux valgus. ГС диагностируется при наличии 2 больших критериев, 1 большого и 2 малых критериев или 4 малых. Достаточно 2 малых критериев, если родственник 1 линии родства имеет признаки ДСТ.
Таблица 2. Диагностические критерии ГС (Brighton, 1998г)
Большие критерии:
Малые критерии:
|
Основным клиническим проявлением ГС является поражение опорно-двигательного аппарата в виде артралгий (полиартралгий), ассоциированных с физической нагрузкой. Наиболее часто в процесс вовлекаются коленные и голеностопные суставы. Причиной болевого синдрома в этом случае является чувствительность к нагрузке опорных суставов и умеренные ортопедические аномалии (дисплазия тазобедренных суставов, продольное и поперечное плоскостопие). Дебют артралгий приходится на молодой возраст, преимущественно у лиц женского пола. Подвывихи суставов (в основном, голеностопных и коленных) типичны для пациентов с ГС. Рецидивирующий выпот в суставе, как проявление ГМС, является нечастой, но наиболее сложной диагностической ситуацией. Характерной особенностью синовита при ГМС является непосредственная связь с травмой, невоспалительный характер синовиальной жидкости и быстрое обратное развитие. В последующему этих пациентов может развиться стойкая артралгия травмированного коленного сустава, связанная с постравматической менископатией. Дорсалгии, нередко сочетающиеся со сколиозом и спондилолистезом, встречаются у пациентов с ГС в любом возрасте. Периартикулярные поражения (тендиниты, эпикондилит, другие энтезопатии, бурситы, туннельные синдромы) встречаются у пациентов с ГС и возникают в ответ на необычную (непривычную) нагрузку или минимальную травму. Наиболее полная картина клинических проявлений и потенциальных осложнений ГС (в том числе со стороны опорно-двигательного аппарата) представлена в табл. 3.
Таблица 3. Клинические проявления и потенциальные осложнения ГС [1]
Острые (травматические)
Хронические (нетравматические)
|
ГМС требует тщательного клинического анализа и дифференцированного диагностического подхода. Немаловажную роль в диагностике ГС играет оценка фенотипических маркеров ДСТ. При наличии сопутствующей гиперрастяжимости кожи (диагностируемой при толщине кожной складки над ключицами > 2 см), атрофичных рубцов, повышенной ранимости кожи, в первую очередь, следует думать о классическом варианте синдрома Элерса-Данло (СЭД) (I/II подтип по классификации Villefranche, 1997). Наличие же врожденного вывиха суставов свидетельствует в пользу артрохалазического типа (VII подтип). Обширные кровоподтеки на коже и/или семейный анамнез сосудистых или кишечных разрывов или внезапной смерти являются признаками сосудистого подтипа СЭД (IV). Если умеренная гипермобильность суставов сочетается с характерным внешним обликом, подвывихом хрусталика и/или дилатацией аорты или аневризмой, необходимо думать о синдроме Марфана. Та же клиническая комбинация без глазных и кардиальных проявлений свидетельствует в пользу гипермобильного подтипа СЭД (III). В настоящее время не определены генетические и нозологические границы между гипермобильным типом СЭД и ГС. Кроме того, было показано, что у пациентов с III подтипом СЭД и ГС имеются мутации в генах, кодирующих неколлагеновые молекулы Tenascin-X, при этом отмечается снижение уровня сывороточного Tenascin-X в обеих группах гетерозиготных лиц женского пола [7]. Идентификация мутаций генов Тenascin-X является важной моделью изучения генетической основы ГС.
Клиническая картина несовершенного остеогенеза характеризуется повышенной ломкостью костей, переломы возникают при небольших нагрузках или спонтанно. Клиническая картина заболевания не ограничивается патологией скелета. Синие склеры, низкий рост, разрушение дентина зубов, прогрессирующая тугоухость в детском и юношеском возрасте, контрактуры, мышечная гипотония и повышенная частота пупочных и паховых грыж, врожденных пороков сердца и нефролитиаза – характерный комплекс патологических изменений при несовершенном остеогенезе. Другие характерные признаки в сочетании с ГМС могут указывать на наличие псевдоахондроплазии (PSACH), синдрома Ларсена, мышечной дистрофии Ульриха и др.
Биохимические и генетические нарушения при ДСТ
Наряду с клинической оценкой ДСТ в диагностике заболевания важную роль играют биохимические методы исследования. Они позволяют оценить состояние обмена соединительной ткани, уточнить диагноз, прогнозировать течение заболевания. Наиболее информативным является определение уровня оксипролина и гликозоаминогликанов в суточной моче, а также лизина, пролина, оксипролина в сыворотке крови. Генетические дефекты синтеза коллагена приводят к уменьшению его поперечных связей и возрастанию количества легкорастворимых фракций. Именно поэтому у больных с врожденной дисплазией соединительной ткани отмечается достоверное повышение оксипролина в суточной моче, степень которого коррелирует с тяжестью патологического процесса. О катаболизме межклеточного вещества судят по величине экскреции гликозоаминогликанов [8].
Для наследственных заболеваний соединительной ткани характерно изменение соотношения коллагенов разных типов и нарушение структуры коллагенового волокна. Типирование коллагена проводится методом непрямой иммунофлюоресценции по Sternberg L.A. при помощи поликлональных антител кфибронектину и коллагену [8]. Современной и перспективной является молекулярно-генетическая диагностика (ДНК-диагностика) дисплазии соединительной ткани, предполагающая применение молекулярных методов выявления генных мутаций. Молекулярный анализ гена fibrillin1 (FBN1) при подозрении на синдром Марфана может быть выполнен на геномной ДНК, извлеченной из лейкоцитов крови. В случаях диагностики СЭД или несовершенного остеогенеза проводится биопсия кожи с последующим биохимическим анализом коллагена типов I, III и V. В зависимости от клинической и биохимической оценки дальнейший молекулярный анализ проводится на ДНК, извлеченной из культивируемых фибробластов.
Алгоритм обследования пациента с гипермобильностью суставов представлен на рис. 1.
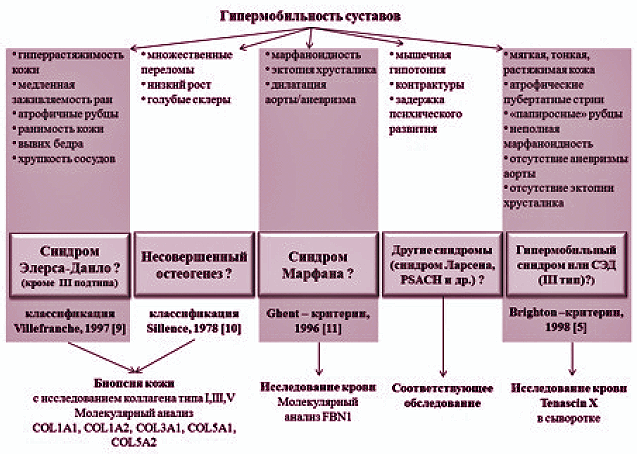
Рисунок 1. Алгоритм диагностики гипермобильности суставов
Возможности лечения ДСТ
Лечение ГМС при отсутствии жалоб не требует специальных мероприятий. При умеренных артралгиях показано ограничение физических нагрузок. Необходимо свести к минимуму возможности травм, что включает профессиональную ориентациюи исключение игровых видов спорта. При упорных болях в одном или нескольких суставах используют эластичные ортезы, обеспечивающие искусственное ограничение объема движений. Немаловажную роль играет укрепление окружающих болезненный сустав мышц с помощью изометрических упражнений, обеспечивающих оптимизацию локальной биомеханики и как следствие – исчезновение болей. В качестве симптоматической медикаментозной терапии при болевом синдроме показан прием нестероидных противовоспалительных препаратов и анальгетиков. С учётом патогенетической основы несостоятельности соединительной ткани и системного характера проявлений ГС основным направлением терапии является коррекция нарушенного метаболизма коллагена. Это позволяет предотвращать возможные осложнения ДСТ. К средствам, стимулирующим коллагенообразование, относят аскорбиновую кислоту, препараты мукополисахаридной природы (хондроитинсульфат, глюкозаминсульфат), витамины группы В (В1, В2, В3, В6) и микроэлементы (медь, цинк, магний). Последние являются кофакторами внутри- и внеклеточного созревания молекулы коллагена и других структурных элементов соединительной ткани [8].
Особая роль в регуляции метаболизма соединительной ткани отводится магнию. В условиях его недостатка происходит усиление деградации коллагеновых и, возможно, эластиновых волокон, а также полисахаридных нитей гиалуронана. Это обусловлено инактивацией гиалуронансинтетаз и эластаз, а также повышением активности гиалуронидаз и матричных металлопротеиназ. На клеточном уровне дефицит магния приводит также к увеличению числа дисфункциональных молекул т-РНК, замедляя, тем самым, скорость белкового синтеза. Кроме того, определенную роль в деградации соединительной ткани играют аутоиммунные реакции, обусловленные присутствием аллеля Bw35 системы HLA. Активация Т-клеточного иммунитета к компонентам соединительной ткани, содержащим рецепторы, соответствующие антигену Вw35, приводит к деградации соединительнотканного матрикса, что сопряжено с неконтролируемой потерей магния [12]. Повышенная экспрессия этого антигена отмечена у пациентов с первичным пролапсом митрального клапана, который является фенотипическим маркером ГС. В ряде исследований показана принципиальная возможность замедления процессов дегенерации соединительной ткани при лечении препаратами магния. Это достигается посредством усиления биосинтетической активности фибробластов, ответственных за нормализацию волокнистых структур соединительнотканного матрикса [13].
Необходимо отметить, что ДСТ имеет, как правило, прогрессирующий характер и лежит в основе формирования соматической патологии. Последняя нередко выходит на первый план и определяет прогноз основного заболевания. Своевременная метаболическая терапия играет важную роль в лечении и профилактике потенциальных осложнений ДСТ, в том числе ГС.
Литература
Комментарии
ПРАКТИКА ПЕДИАТРА



