Секреты патогенеза целиакии
СтатьиЮ.А. Дмитриева1, 2, канд. мед. наук, С.Е. Тесленко3, Л.С. Абдурахманова4, И.М. Османов2, д-р мед. наук, профессор,И.Н. Захарова1, д-р мед. наук, профессор
1 ФГБОУ ДПО «Российская медицинская академия непрерывного профессионального образования» Минздрава России, г. Москва
2 ГБУЗ «Детская городская клиническая больница им. З.А. Башляевой Департамента здравоохранения г. Москвы»
3 ФГАОУ ВО «Российский национальный исследовательский медицинский университет им. Н.И. Пирогова» Минздрава России, г. Москва
4 ФГБУ «Детский медицинский центр» Управления делами Президента РФ, г. Москва
Резюме. В основе патогенеза целиакии лежит патологический процесс, опосредованный клеточными и гуморальными компонентами врожденного и адаптивного иммунного ответа. Обязательными факторами, определяющими развитие глютеновой энтеропатии, является регулярное употребление глютенсодержащих продуктов и наличие генетической предрасположенности, которая определяется присутствием у пациентов генов главного комплекса гистосовместимости HLA-DQ2/DQ8. Центральным событием патогенеза заболевания является связывание пептидов глиадина с HLA-DQ2/DQ8-молекулами с последующей презентацией их глютенспецифическим CD4+ Т-лимфоцитам и развитием воспалительного процесса в слизистой оболочке тонкой кишки с формированием атрофической энтеропатии и разнообразных клинических проявлений заболевания.
Ключевые слова: целиакия, глютен, зонулин, тканевая трансглутаминаза, главный комплекс гистосовместимости, молекулы HLA-DQ2/DQ8, инфекция, микробиота, сроки введения глютена, грудное вскармливание, врожденный и адаптивный иммунный ответ, цитокины, дендритные клетки,межэпителиальные лимфоциты, энтеропатия
Summary. The pathogenesis of celiac disease is based on a pathological process mediated by cellular and humoral components of the innate and adaptive immune response. Mandatory factors determining the development of gluten enteropathy are regular consumption of gluten-containing products and the presence of a genetic predisposition, which is determined by the presence of genes of the main histocompatibility complex HLA-DQ2/DQ8 in patients. The central event in the pathogenesis of the disease is the binding of gliadin peptides to HLA-DQ2/DQ8 molecules, followed by their presentation to gluten-specific CD4+ T lymphocytes and the development of an inflammatory process in the mucous membrane of the small intestine with the formation of atrophic enteropathy and various clinical manifestations of the disease.
Keywords: celiac disease, gluten, zonulin, tissue transglutaminase, major histocompatibility complex, HLA-DQ2/DQ8 molecules, infection, microbiota, timing of gluten administration, breastfeeding, innate and adaptive immune response, cytokines, dendritic cells, interepithelial lymphocytes, enteropathy
Для цитирования: Секреты патогенеза целиакии / Ю.А. Дмитриева [и др.] // Практика педиатра. 2024. № 4. С. 30-36.
For citation: Secrets of the pathogenesis of celiac disease / Yu.A. Dmitrieva [et al.] // Pediatrician's practice. 2024;(4): 30-36. (in Russ.)
Целиакия представляет собой иммуноопосредованное системное заболевание, которое возникает в ответ на употребление глютена генетически предрасположенными лицами и характеризуется наличием широкой комбинации глютен-зависимых симптомов, специфических антител (антител к тканевой трансглутаминазе, эндомизию и деамидированым пептидам глиадина), наличием HLA-DQ2 или HLA-DQ8 гаплотипов и энтеропатии [1,2]. В определении целиакии заложены основные представления о патогенезе заболевания. Обязательными факторами, определяющими развитие глютеновой энтеропатии, являются регулярное употребление глютенсодержащих продуктов и наличие генетической предрасположенности к заболеванию.
Глютен составляет до 80% белковой фракции пшеницы, ржи и ячменя. В структуре этих злаков данный белковый компонент имеет собственное название (глиадины пшеницы, секалины ржи, хордеины ячменя), при этом в медицинской литературе указанные белки объединены общим названием «глютен» с учетом филогенетического и структурного сходства, а также аналогичного воздействия на организм человека [3]. Глиадины пшеницы богаты пролином и глутамином, что делает их устойчивыми к воздействию желудочных, панкреатических и интестинальных протеиназ и определяет сохранение их высокого иммуногенного потенциала при прохождении через желудочно-кишечный тракт человека [4]. Основным инициатором иммуновоспалительного ответа в слизистой кишечника признан 33-мерный пептид LQLQPFPQPQLPYPQPQLPYPQPQLPYPQPQPH, выделенный в составе рекомбинантного а2-глиадина в 2002 г. [5]. Наряду с высокой устойчивостью глютена к энзиматическому воздействую, его структура уникальна еще и тем, что он содержит определенные пептидные фрагменты, облегчающие его проникновение через эпителиальный барьер кишечника (рис. 1). В регуляции проницаемости кишечника важную роль играет зонулин - белок группы гаптоглобинов, вырабатываемый в печени и тканях внутреннего эпителия, являющийся главным модулятором тесных белковых соединений в межклеточном пространстве. В настоящее время доказано, что отдельные эпитопы глютена являются активаторами экспрессии зонулина, нарушая тем самым тесные связи между энтероцитами. Указанные изменения приводят к избыточному проникновению различных антигенов, включая сам глютен, во внутреннюю среду организма и, как следствие, к активации иммунной системы и продукции комплекса провоспалительных цитокинов [6].
 Рис. 1. Молекулярная структура глютена (адаптировано из [6])
Рис. 1. Молекулярная структура глютена (адаптировано из [6])
Употребление в пищу глютенсодержащих продуктов является необходимым, но недостаточным условием развития патологического процесса в слизистой оболочке тонкой кишки. Целиакия является генетически-детерминированным заболеванием, в патогенезе которого важную роль играют гены, входящие в главный комплекс гистосовместимости человека [7]. У человека данный комплекс носит также название HLA (Human Leukocyte Antigen complex). Главный комплекс гистосовместимости представляет собой группу генов и кодируемых ими антигенов (молекул) клеточной поверхности, которые играют важнейшую роль в распознавании чужеродных белков и развитии иммунного ответа. HLA располагается на коротком плече 6 хромосомы, занимая регион размером 4Mb (около тысячи пар оснований), и содержит более 200 генов. Основная функция системы HLA заключается в регуляции иммунного ответа путем генетического контроля взаимодействия иммунокомпетентных клеток организма. Индивидуальный набор и свойства молекул HLA во многом определяют силу иммунного ответа конкретного человека на конкретный антиген [8, 9]. В патогенезе целиакии участвуют гены локуса DQ в составе HLA II класса, а в частности гены DQ2 и DQ8 [10]. Антигены (молекулы), кодируемые HLA-DQ2/DQ8, экспрессируются на поверхности ангигенпрезентирующих клеток [9] и представляют собой гетеродимеры - белки, состоящие из двух цепей (альфа и бета). Роль данных молекул в патогенезе целиакии обусловлена их участием в процессе презентации пептидов глиадина CD4+ лимфоцитам.
Следует отметить, что гетеродимеры DQ2/DQ8 встречаются в популяции с частотой 30%, однако частота целиакии, в соответствии с современными эпидемиологическими исследованиями, составляет 1%. Принято считать, что антигены HLA-DQ2 или HLA-DQ8 определяют риск развития заболевания лишь 36-53% [11]. Поиск других возможных генетических факторов, ответственных за его развитие, продолжается до настоящего времени. Проведенные исследования позволили установить возможную связь развития целиакии с не HLA-генами, расположенными на 5 (5q31-33), 2 (2q33), 19 (19p13), 4 (4q27) хромосомах [12]. Гены указанных локусов играют важную роль в осуществлении регуляции продукции цитокинов (TNFa, IFNy, IL 2, IL 21, IL 10) и активации естественных киллеров, Т- и В-лимфоцитов, а также в поддержании барьерной функции слизистой оболочки тонкой кишки [13, 14]. Мутации в данных регуляторных участках часто выявляются у пациентов с воспалительными заболеваниями кишечника, инсулинозависимым сахарным диабетом, системной красной волчанкой, ревматоидным артритом и др., что раскрывает одну из возможных причин частой ассоциации целиакии с данными патологическими состояниями [15, 16].
С момента формирования современных представлений о патогенезе целиакии стала активно обсуждаться роль средовых факторов в развитии заболевания. Интерес к данной проблеме был в том числе обусловлен возможностью разработки профилактических мероприятий в группах риска, способных отсрочить дебют заболевания или предотвратить развитие его тяжелых манифестных форм. Наибольшее количество исследований среди детского населения посвящено влиянию характера вскармливания в младенческом и раннем возрасте на риск развития целиакии в целом и возраст дебюта заболевания. Результаты крупных систематических обзоров и метаанализов клинических исследований, выполненных в первом десятилетии XXI в., указали на протективный эффект продолжительного естественного вскармливания, а также грудного молока в момент первого контакта ребенка с глютеном, в отношении раннего дебюта заболевания, хотя не все результаты исследований выглядели однозначными [17, 18]. Многие исследователи указывали на существование оптимального периода знакомства младенца с глютеном, когда возможно формирование толерантности к данному белку, что, соответственно, могло привести к снижению риска реализации генетической предрасположенности к целиакии [19, 20]. Доступные результаты исследований послужили основанием для публикации официальных рекомендаций Европейского общества детских гастроэнтерологов, гепатологов и нутрициологов (ESPGHAN) в отношении оптимального срока введения глютенсодержащих продуктов прикорма в рацион ребенка первого года жизни, определив последний как 4-6 мес, что связывалось с возможностью профилактики аллергии к злакам и целиакии [21]. В 2007 г. стартовал проект PreventCD, представляющий собой крупное проспективное рандомизированное двойное слепое интервенционное мультицентровое исследование [22]. Исследование было проведено в восьми странах и включило более 900 детей из группы риска, являющихся носителями HLA-DQ2/DQ8 гаплотипов. В возрасте до 3 мес участники исследования были рандомизированы на две группы в зависимости от особенностей введения продуктов прикорма. Младенцы основной группы в период с 16-й недели жизни на протяжении последующих двух месяцев получали глютен, в то время как дети группы контроля - плацебо. При оценке частоты целиакии в возрасте 3 лет было установлено, что частота целиакии достоверно не отличалась у детей из обеих групп. Дополнительная оценка роли грудного вскармливания продемонстрировала отсутствие достоверного протективного эффекта грудного молока в отношении развития заболевания. В качестве основных факторов риска развития целиакии авторами были установлены принадлежность к женскому полу и особенности HLA-генотипа. Так, заболевание развивалось достоверно более часто и в более раннем возрасте у DQ2 гомозигот [22]. Аналогичные результаты были получены в исследовании CELIPREV, выполненного итальянскими авторами [23]. Исследователи продемонстрировали, что основным фактором риска раннего дебюта развития заболевания является присутствие в генотипе аллелей высокого риска (двойной копии молекулы DQ2), в отсутствии достоверного влияния грудного вскармливания и сроков введения глютена в рацион питания младенца.
Среди возможных триггерных факторов различными исследователями рассматривались особенности течения беременности у матери и осложнения неонатального периода [24], перенесенные ребенком вирусные кишечные инфекции [25, 26]. Вопрос о роли инфекционных агентов в качестве триггеров целиакии до сих пор остается нерешенным. В различных исследованиях было подтверждено участие энтеровирусных, ротавирусных и других желудочно-кишечных инфекций в модуляции иммунного ответа при целиакии, которые при этом повышают риск этого заболевания [27, 28]. Кишечная реовирусная инфекция индуцировала иммунный ответ при пероральном употреблении глютена. Утрату толерантности связывали с индукцией IFN, что способствовало активации антиглютенового ответа T- и B-клеток и появлению антител против тканевой трансглутаминазы [29]. В некоторых исследованиях было показано снижение заболеваемости у детей, вакцинированных против ротавирусной инфекции [30], но это не было подтверждено при эксперименте на большей когорте пациентов [31].
Многочисленные исследования зафиксировали различия в микробиоте пациентов с целиакией по сравнению с контрольной группой с увеличением количества протеобактерий и уменьшением количества штаммов, обладающих противовоспалительными свойствами, что обнаруживается при многих хронических заболеваниях кишечника [32, 33]. Однако интересно, что микробы могут по-разному влиять на деградацию глютена в просвете кишечника и либо способствовать высвобождению высокоиммуногенных пептидов (например, Pseudomonas aeruginosa), либо, наоборот, снижать их просветную концентрацию (Lactobacillus) [34]. Кроме того, была обнаружена перекрестная активация белков глютена с большим спектром пептидов комменсальных бактерий [35]. Так, например, против пептидов, полученных из Pseudomonas fluorescens, 86% пациентов с целиакией дают положительный результат при серологической диагностике [36]. Тем не менее не исключено, что Т-клетки, активированные глютеном, могут случайным образом перекрестно реагировать с бактериальными пептидами. Кроме того, поскольку титры антител против этих пептидов сопоставимы у лиц, находящихся на безглютеновой диете и не соблюдающих ее, тот факт, что активность заболевания значительно снижается на фоне безглютеновой диеты, говорит в пользу того, что присутствие бактерий, несущих мимикрирующие пептиды, недостаточно для поддержания воспаления [37].
В основе патогенеза целиакии лежат сложные взаимодействия между глютеном, тканевой трансглутаминазой, врожденной и адаптивной иммунной системами, что в конечном итоге приводит к формированию атрофической энтеропатии и разнообразным клиническим проявлениям заболевания.
Последовательности, богатые пролином и глутамином, в пептидах глютена являются мишенями для фермента тканевой трансглутаминазы 2 типа, играющей важнейшую роль в патогенезе глютеновой энтеропатии. Трансглутаминазы представляют собой кальций-зависимые ферменты, которые катализируют перекрестное сшивание белков, а также могут превращать остатки глутамина в глутаминовую кислоту посредством гидролиза (деамидирования) [38]. Считается, что тТГ экспрессируется повсеместно и участвуют в ремоделировании тканей посредством перекрестного связывания белков внеклеточного матрикса. Однако распространенность тТГ во внеклеточной среде остается неопределенной. Многочисленные исследования показали, что тТГ не распространена во внеклеточной среде при нормальных условиях, а дезамидирование, опосредованное внеклеточной тТГ, индуцируется лишь во время воспаления тканей [39]. Поскольку тТГ высоко экспрессируется в энтероцитах, было высказано предположение, что при целиакии этот фермент высвобождается из погибающих энтероцитов с последующим поглощением из просвета кишки [40].
Тканевая трансглутаминаза осуществляет дезамидирование нейтральных остатков глутамина, превращая их в отрицательно заряженную глутаминовую кислоту [41]. В ходе этой ферментативной реакции происходит повышение афинности пептидов глютена к связывающим участкам молекул DQ2 и DQ8 на поверхности антигенпрезентирующих клеток (АПК), способствуя эффективной активации глютен-специфичных [42]. После примирования в связанной с кишечником лимфоидной ткани глютен-специфичные CD4+ Т-лимфоциты мигрируют в собственную пластинку тонкой кишки и продуцируют провоспалительные цитокины, повреждающие энтероциты, такие как IFNy, IL-21, IL-10, IL-2, а также стимулируют B-клетки к продукции специфических антител (рис. 2).
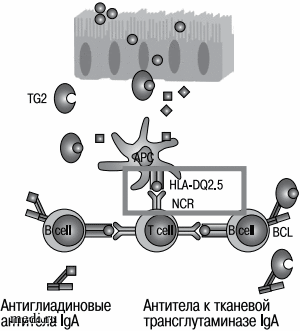 Рис. 2. Патогенез целиакии (адаптировано из [43])
Рис. 2. Патогенез целиакии (адаптировано из [43])
В исследовании Goel et al. пероральное введение глютена вызвало значительное повышение уровня IL-2 в плазме в течение четырех часов, что указывает на быструю активацию Т-лимфоцитов [44]. Многие пептиды глютена являются чрезвычайно хорошими субстратами для тТГ, позволяя образовывать прочные комплексы тТГ-глютен, что вызывает активацию тТГ-специфичных В-клеток и, как следствие, приводит к образованию тТГ-специфичных аутоантител [38]. После активации специфические к глютену CD4+ Т-лимфоциты взаимодействуют с наивными глютен-специфичными В-клетками, индуцируя гуморальный ответ. Кроме того, комплексы тТГ-глютен могут проникать через эпителий пейеровой бляшки и связываться с В-клеточным рецептором аутореактивных анти-тТГ В-лимфоцитов, присутствующих в лимфоидном фолликуле. Комплексы тТГ-глютен, связанные с рецептором В-клеток, подвергаются эндоцитозу и процессингу в лизосомах В-лимфоцитов, где пептиды глютена высвобождаются и формируют комплексы с HLA-DQ, которые затем транслоцируются на поверхность аутореактивных В-клеток, позволяя активировать глютен-специфичные CD4 + Т-клетки. При этом одновременно осуществляются кооперативные взаимодействия между кланами лимфоцитов, которые запускают выработку IgA. При повторной встрече с комплексами HLA-DQ-глютен, представленными АПК, эффекторные CD4+ Т-клетки секретируют провоспалительные цитокины, а также участвуют в активации CD8+ цитотоксических Т-межэпителиальных лимфоцитов и плазматических клеток, которые вырабатывают антитела как к глютену, так и к тканевой трансглутаминазе тТГ [41]. Активация цитотоксических CD8+ Т-клеток происходит благодаря взаимодействию между цитокинами, высвобождаемыми активированными глютен-специфичными CD4+ Т-клетками, и IL-15, продуцируемым эпителиальными и миелоидными клетками при активном воспалении. Эти T-лимфоциты вызывают аутолиз эпителиальных клеток слизистой оболочки тонкой кишки [45]. Цитотоксические CD8+ T-клетки синтезируют IFNy и цитотоксические молекулы, такие как гранзим B и перфорин, а также повышают активность активирующих рецепторов естественных киллеров NKG2D и CD94/NKG2C, тем самым облегчая взаимодействие цитотоксических CD8+ T-клеток с энтероцитами, которые при активном течении целиакии в ответ на тканевой стресс и IFNy экспрессируют MICA (лиганд NKG2D) и HLA-E (лиганд NKG2C) [41].
Собственная пластинка слизистой оболочки тонкой кишки содержит самую большую популяцию плазматических клеток во всем организме. Большинство этих клеток секретируют Ig класса А, которые высвобождаются в просвет кишечника, где они могут связываться с бактериями и пищевыми антигенами [38]. Используя поверхностную экспрессию B-клеточного рецептора плазматическими клетками, были проведены исследования, показавшие, что на тТГ отвечают 10-20% этих клеток, в то время как лишь 1% реагирует на ДПГ [46-48]. Таким образом, идентификация антигенспецифических плазматических клеток указывает на то, что реакция B-клеток в кишечнике сильно смещена в сторону образования антител против тТГ, а не против глютена. Кроме того, было показано, что сывороточные IgA вырабатываются кишечными плазматическими клетками, в то время как сывороточные IgG не секретируются IgG-плазматическими клетками кишечника и, вероятно, экспрессируются в дренирующих тонкую кишку мезентериальных лимфатических узлах, которые также подвергаются действию пептидов глютена и тТГ [46].
Вопрос о том, играют ли аутоантитела какую-либо патогенную роль при целиакии, остается нерешенным. В исследованиях было показано, что при введении мышам полученных от пациента специфичных к тТГ антител могут возникать незначительные морфологические изменения в тонком кишечнике, однако они не вызывают состояния, напоминающего целиакию [49], а экспрессия тТГ-специфических моноклональных антител in vivo у мышей не вызывала заболевания [50]. Это позволяет сделать вывод, что антитела IgA при целиакии не являются непосредственно патогенными, что также подтверждается фактом об увеличении риска развития целиакии до 15 раз у людей с селективным дефицитом IgA [51]. Однако, помимо секреции антител, экспрессия мембраносвязанного B-клеточного рецептора делает В-клетки эффективными АПК для Т-клеток благодаря их способности специфически связывать и представлять антиген. Во время иммунного ответа антигенспецифические В-клетки могут быстро являться наиболее распространенными АПК для CD4+ Т-клеток [38]. При этом Т- и В-клетки активируют дальнейшую клональную экспансию, что приводит к дифференцировке как В-клеток в плазматические клетки, так и Т-клеток в эффекторные. Важность таких взаимодействий B- и T-лимфоцитов подтверждается исследованием Lеjеunеу еt al., при котором на мышиной модели было показано, что истощение B-клеток предотвращает накопление патогенных Т-клеток и развитие активной формы заболевания [52].
Помимо повышенного количества Т-клеток и плазматических клеток у пациентов с активной целиакией, описано увеличение популяций клеток врожденного иммунного ответа, включая эозинофилы, базофилы, тучные клетки, нейтрофилы и дендритные клетки [43, 53]. Увеличение числа тучных клеток при активной целиакии коррелирует с гистологической оценкой Marsh и становится клеточным источником TNFa, IE-6, IE-17, а также моноцитарного хемоаттрактантного белка 1 [53, 54]. Нейтрофильная инфильтрация тонкой кишки в ответ на введение глютена может быть объяснена секрецией IE-8 активированными T-клетками [55]. Также было обнаружено, что пептиды глиадина обладают свойствами хемоаттрактанта нейтрофилов [56]. Более того , даже у пациентов в стадии ремиссии целиакии отмечалась инфильтрация нейтрофилами слизистой тонкой кишки [57]. В другом исследовании после введения глютена наблюдалось увеличение плотности нейтрофилов, а также моноцитов и дендритных клеток [58]. Дендритные клетки могут выполнять различные функции, такие как индукция воспалительной реакции, стимулирование Т-клеток и контроль иммунного ответа путем индукции регуляторных Т-клеток [59, 60].
С патогенетической точки зрения целиакия является одним из наиболее изученных иммуноопосредованных заболеваний, однако многое в механизмах ее развития еще предстоит исследовать. Существующие представления о патогенезе глютеновой энтеропатии положены в основу современного протокола диагностики, а также являются неотъемлемой частью процесса разработки методов медикаментозного лечения заболевания.
Литература
1. Целиакия у детей: проект клинических рекомендаций / Е.А. Рославцева [и др.] // Экспериментальная и клиническая гастроэнтерология. 2021. № 188 (4). С. 199-227.
2. ESPGHAN Working Group on Coeliac Disease Diagnosis; ESPGHAN Gastroenterology Committee; European Society for Pediatric Gastroenterology, Hepatology, and Nutrition. European Society for Pediatric Gastroenterology, Hepatology, and Nutrition guidelines for the diagnosis of coeliac disease / S. Husby [et al.] // Journal of Pediatric Gastroenterology and Nutrition. 2012. Vol. 54, No. 1. P 136-160.
3. Целиакия: клинические особенности / И.Н. Захарова [и др.] // Педиатрия. Consilium Medicum. 2014. № 3. С. 62-67.
4. Spectrum of gluten-related disorders: consensus on new nomenclature and classification / A. Sapone [et al.] // BMC Medicine. 2012. Vol. 7, No. 10. P. 13.
5. Structural basis for gluten intolerance in celiac sprue / L. Shan [et al.] // Science. 2002. Sep. 27. Vol. 297, No. 5590. P 22752279.
6. Fasano A. Zonulin and its regulation of intestinal barrier function: the biological door to inflammation, autoimmunity, and cancer // Physiological Reviews. 2011. Vol. 91, No. 1. P. 151175.
7. Структура HLA-DR-DQ-генотипа у детей с целиакией / Ю.А. Дмитриева [и др.] // Медицинский совет. 2020. № 10. С. 74-80.
8. Genetics in coeliac disease / D.A. van Heel [et al.] // Best Practice & Research Clinical Gastroenterology. 2005. Vol. 19, No. 3. P 323-339.
9. Дранник Г.Н. Клиническая иммунология и аллергология. М. : ООО «Медицинское информационное агентство», 2003. 604 с.
10. Sollid L.M. Molecular basis of celiac disease // Annual Review of Immunology. 2000. Vol. 18. P. 53-81.
11. Genetic contribution of the HLA region to the familial clustering of coeliac disease / F. Petronzelli [et al.] // Annals of Human Genetics. 1997. Vol. 61 (Pt 4). P 307-317.
12. Catassi C., Fasano A. Celiac disease // Curr Opin Gastroenterol. 2008. Vol. 24, No. 6. C. 687-691.
13. Candidate gene regions and genetic heterogeneity in gluten sensitivity / P. Holopainen [et al.] // Gut. 2001. Vol. 48, No. 5. P 696-701.
14. Evaluation of cytokine polymorphisms (TNFalpha, IFNgamma and IL-10) in Down patients with coeliac disease / F. Cataldo [et al.] // Digestive and Liver Disease. 2005. Vol. 37, No. 12. P 923-927.
15. Epidemic of coeliac disease in Swedish children / A. Ivarsson [et al.] // Acta Paediatrica. 2000. Vol. 89, No. 2. P. 165-171.
16. Целиакия и ассоциированные эндокринные заболевания / Ю.А. Дмитриева, И.Н. Захарова, И.М. Османов [и др.] // Практика педиатра. 2022. № 2. С. 23-31.
17. Effect of breast feeding on risk of coeliac disease: a systematic review and meta-analysis of observational studies / A.K. Ako-beng [et al.] // Archives of Disease in Childhood. 2006. Vol. 91, No. 1. P 39-43.
18. Henriksson C., Bostrom A.M., Wiklund I.E. What effect does breastfeeding have on coeliac disease? A systematic review update // Evidence-based medicine (EBM). 2013. Vol. 18, No. 3. P 98-103.
19. Risk of celiac disease autoimmunity and timing of gluten introduction in the diet of infants at increased risk of disease / J.M. Norris [et al.] // JAMA. 2005. May 18. Vol. 293, No. 19. P 2343-2351.
20. Stordal K., White R.A., Eggesbo M. Early feeding and risk of celiac disease in a prospective birth cohort // Pediatrics. 2013. Vol. 132, No. 5. P e1202-9.
21. Complementary feeding: a commentary by the ESPGHAN Committee on Nutrition / C. Agostoni [et al.] // Journal of Pediatric Gastroenterology and Nutrition. 2008. Vol. 46, No. 1. P 99-110.
22. Randomized feeding intervention in infants at high risk for celiac disease / S.L. Vriezinga [et al.] // New England Journal of Medicine (NEJM). 2014. Oct. 2. Vol. 371, No. 14. P 1304-1315.
23. Introduction of gluten, HLA status, and the risk of celiac disease in children / E. Lionetti [et al.] // New England Journal of Medicine (NEJM). 2014. Oct. 2. Vol. 371, No. 14. P 1295-1303.
24. Sandberg-Bennich S., Dahlquist G., Kallen B. Coeliac disease is associated with intrauterine growth and neonatal infections // Acta Paediatrica. 2002. Vol. 91, No. 1. P. 30-33.
25. Plot L., Amital H. Infectious associations of Celiac disease // Autoimmunity Reviews. 2009. Vol. 8, No. 4. P. 316-319.
26. Rotavirus infection frequency and risk of celiac disease autoimmunity in early childhood: a longitudinal study / L.C. Stene [et al.] // American Journal of Gastroenterology. 2006. Vol. 101, No. 10. P 2333-2340.
27. Enterovirus as trigger of coeliac disease: nested case-control study within prospective birth cohort / C.R. Kahrs [et al.] // British Medical Journal. 2019. Feb. 13. Vol. 364. P. l231.
28. Enterovirus Infections Are Associated With the Development ofCeliac Disease in a Birth Cohort Study / M. Oikarinen [et al.] // Frontiers In Immunology. 2021. Feb. 2. Vol. 11. P. 604529.
29. Reovirus infection triggers inflammatory responses to dietary antigens and development of celiac disease / R. Bouziat [et al.] // Science. 2017. Apr. 7. Vol. 356, No. 6333. P 44-50.
30. Rotavirus Vaccination Does Not Increase Type 1 Diabetes and May Decrease Celiac Disease in Children and Adolescents / M. Hemming-Harlo [et al.] // Pediatric Infectious Disease Journal. 2019. Vol. 38, No. 5. P 539-541.
31. Paediatric rotavirus vaccination, coeliac disease and type 1 diabetes in children: a population-based cohort study / T. Inns [et al.] // BMC Medicine. 2021. Jun. 29. Vol. 19, No. 1. P 147.
32. Микробиота и дисбиоз кишечника при целиакии / С.И. Ситкин, Е.Б. Авалуева, Л.С. Орешко, А. Хавкин // Российский вестник перинатологии и педиатрии. 2021. Т. 66, № 2. C. 116-122.
33. Verdu E.F., Schuppan D. Co-factors, Microbes, and Immunogenetics in Celiac Disease to Guide Novel Approaches for Diagnosis and Treatment // Gastroenterology. 2021. Vol. 161, No. 5. P 1395-1411.e4.
34. Duodenal Bacteria From Patients With Celiac Disease and Healthy Subjects Distinctly Affect Gluten Breakdown and Immunogenicity / A. Caminero [et al.] // Gastroenterology. 2016. Oct. Vol. 151, No. 4. P 670-683.
35. T cell receptor cross-reactivity between gliadin and bacterial peptides in celiac disease / J. Petersen [et al.] // Nature Structural & Molecular Biology. 2020. Vol. 27, No. 1. P. 49-61.
36. Serological responses to microbial antigens in celiac disease patients during a gluten-free diet / S. Ashorn [et al.] // Journal of Clinical Immunology. 2009. Vol. 29, No. 2. P. 190-195.
37. Microbial Biomarkers in Patients with Nonresponsive Celiac Disease / L. Viitasalo [et al.] // Digestive Diseases and Sciences. 2018. Vol. 63, No. 12. P. 3434-3441.
38. Iversen R., Sollid L.M. The Immunobiology and Pathogenesis of Celiac Disease // Annual Review of Pathology: Mechanisms of Disease. 2023. Vol. 24, No. 18. P. 47-70.
39. IL-15, gluten and HLA-DQ8 drive tissue destruction in coeliac disease / V Abadie [et al.] // Nature. 2020. Vol. 578, No. 7796. P. 600-604.
40. Evidence That Pathogenic Transglutaminase 2 in Celiac Disease Derives From Enterocytes / R. Iversen [et al.] // Gastroenterology. 2020. Vol. 159, No. 2. P. 788-790.
41. Levescot A., Malamut G., Cerf-Bensussan N. Immunopathogenesis and environmental triggers in coeliac disease // Gut. 2022. Jul. 25. Vol. 71, No. 11. P. 2337-2349.
42. Sollid L.M. The roles of MHC class II genes and post-translational modification in celiac disease // Immunogenetics. 2017. Vol. 69, No. 8-9. P. 605-616.
43. Sollid L.M., Jabri B. Celiac disease and transglutaminase 2: a model for posttranslational modification of antigens and HLA association in the pathogenesis of autoimmune disorders // Current Opinion in Immunology. 2011. Vol. 23, No. 6. P. 732738.
44. Cytokine release and gastrointestinal symptoms after gluten challenge in celiac disease / G. Goel [et al.] // Science Advances. 2019. 7. Vol. 5, No. 8. P. eaaw7756.
45. Waldmann T.A., Miljkovic M.D., Conlon K.C. Interleukin-15 (dys)regulation of lymphoid homeostasis: Implications for therapy of autoimmunity and cancer // Journal of Experimental Medicine. 2020. Jan. 6. Vol. 217, No. 1. P. :e20191062.
46. Strong Clonal Relatedness between Serum and Gut IgA despite Different Plasma Cell Origins / R. Iversen [et al.] // Cell Reports. 2017. Sep. 5. Vol. 20, No. 10. P. 2357-2367.
47. Longevity, clonal relationship, and transcriptional program of celiac disease-specific plasma cells / I. Lindeman [et al.] // Journal of Experimental Medicine. 2021. Feb. 1. Vol. 218, No. 2. P. :e20200852.
48. Restricted VH/VL usage and limited mutations in gluten-specific IgA of coeliac disease lesion plasma cells / 0. Dunand [et al.] // Nature Communications. 2014. Vol. 9, No. 5. P. 4041.
49. Transglutaminase 2-specific coeliac disease autoantibodies induce morphological changes and signs of inflammation in the small-bowel mucosa of mice / S. Kalliokoski [et al.] // Amino Acids. 2017. Vol. 9, No. 3. P. 529-540.
50. B cell tolerance and antibody production to the celiac disease autoantigen transglutaminase 2 / M.F. du Pre [et al.] // Journal of Experimental Medicine. 2020. Feb. 3. Vol. 217, No. 2. P. :e20190860.
51. Pediatric Celiac Disease and Selective IgA Deficiency: Unexpected Sequence of Events / M.C.E. Andersen [et al.] // Journal of Clinical Immunology. 2022. Aug. Vol. 42, No. 6. P. 13421346.
52. Lejeune T., Meyer C., Abadie V. B Lymphocytes Contribute to Celiac Disease Pathogenesis // Gastroenterology. 2021. Vol. 160, No. 7. P. 2608-2610.e4.
53. Mast cells are associated with the onset and progression of celiac disease / B. Frossi [et al.] // Journal of Allergy and Clinical Immunology. 2017. Vol. 139, No. 4. P. 1266-1274.e1.
54. Marsh M.N., Heal C.J. Evolutionary Developments in Interpreting the Gluten-Induced Mucosal Celiac Lesion: An Archimedian Heuristic // Nutrients. 2017. Vol. 9, No. 3. P. 213.
55. Cytokine release and gastrointestinal symptoms after gluten challenge in celiac disease / G. Goel [et al.] // Science Advances. 2019. Aug. 7. Vol. 5, No. 8. P. :eaaw7756.
56. Gliadin Induces Neutrophil Migration via Engagement of the Formyl Peptide Receptor, FPR1 / K.M. Lammers [et al.] // PLoS One. 2015. Sep. 17. Vol. 10, No. 9. P. :e0138338.
57. Neutrophil recruitment and barrier impairment in celiac disease: a genomic study / B. Diosdado [et al.] // Clinical Gastroenterology and Hepatology. 2007. Vol. 5, No. 5. P. 574-581.
58. Rapid accumulation ofCD14+CD11c+ dendritic cells in gut mucosa of celiac disease after in vivo gluten challenge / A.C. Beitnes [et al.] // PLoS One. 2012. Vol. 7, No. 3. P. :e33556.
59. Anderson R.P. Innate and adaptive immunity in celiac disease // Curr Opin Gastroenterol. 2020. Vol. 36, No. 6. P. 470-478.
60. Coeliac Disease Pathogenesis: The Uncertainties of a Well-Known Immune Mediated Disorder / M.R. Dunne [et al.] // Frontiers in Immunology. 2020. Vol. 8, No. 11. P. 1374.



