Структура задержки полового созревания у девочек
СтатьиК.Л. Каболова1, О.Ю. Латышев1, канд. мед. наук, Г.Ф. Окминян1, канд. мед. наук, Е.В. Киселева1, канд. мед. наук, Д.С. Ромайкина1, Л.Н. Самсонова1, д-р мед. наук, профессор, И.М. Османов2, 3, д-р мед. наук, профессор
1 ФГБОУ ДПО «Российская медицинская академия непрерывного профессионального образования» Минздрава России, г. Москва
2 ГБУЗ «Детская городская клиническая больница им. З.А. Башляевой Департамента здравоохранения Москвы»
3 ФГАОУ ВО «Российский национальный исследовательский медицинский университет им. Н.И. Пирогова» Минздрава России, г. Москва
Резюме. Исследование посвящено изучению структуры задержки полового созревания у девочек в зависимости от этиологии и клинической картины. Анализ результатов проведенного исследования показал, что структура задержки полового созревания у девочек в равной степени представлена перманентной и транзиторной формами. В перманентной форме значимо преобладает гипергонадотропный гипогонадизм, тогда как гипогонадотропный гипогонадизм встречается в 1/6 случаев. Среди пациенток с транзиторной формой задержки полового созревания конституциональная задержка пубертата и функциональный гипогонадизм встречались с одинаковой частотой. Причиной обращения девочек с перманентной формой чаще являлась первичная аменорея, тогда как в группе с транзиторной формой - отсутствие вторичных половых признаков. Знание структуры задержки полового созревания помогает понять причину данного состояния и выбрать правильную траекторию диагностического поиска, что способствует более быстрой постановке диагноза и выбора дальнейшей тактики ведения.
Ключевые слова: гипергонадотропный гипогонадизм, гипогонадотропный гипогонадизм, девочки, функциональный гипогонадизм, конституциональная задержка пубертата
Summary. The study is devoted to studying the structure of delayed puberty in girls depending on the etiology and clinical picture. Analysis of the results of the study showed that the structure of delayed puberty in girls is equally represented by permanent and transient forms. In the permanent form, hypergonadotropic hypogonadism significantly predominates, while hypogonadotropic hypogonadism occurs in 1/6 of cases. Among patients with a transient form of delayed puberty, constitutional delayed puberty and functional hypogonadism occurred with equal frequency. The reason for treatment of girls with the permanent form was more often primary amenorrhea, while in the group with the transient form it was the absence of secondary sexual characteristics. Knowledge of the structure of delayed puberty helps to understand the cause of this condition and choose the correct trajectory of the diagnostic search, which contributes to a faster diagnosis and choice of further management tactics.
Keywords: hypergonadotropic hypogonadism, hypogonadotropic hypogonadism, girls, functional hypogonadism, constitutional delayed puberty
Для цитирования: Структура задержки полового созревания у девочек / К.Л. Каболова [и др.] // Практика педиатра. 2024. № 3. С. 30-36.
For citation: The structure of girls' delayed puberty / K.L. Kabolova [et al.] // Pediatrician's practice. 2024;(3): 30-36. (in Russ.)
Половое созревание - важный период физического, психологического и социального развития человека. Это пролонгированный во времени процесс, состоящий из серии сложных событий, которые начинаются внутриутробно, активируются в неонатальном периоде, а затем, после относительного покоя в детстве, реактивируются в начале пубертатного развития. Процесс полового созревания рассматривается как «биологический сенсор», а изменения в его сроках служат потенциальным биомаркером соматического и психического здоровья человека, а также негативного влияния окружающей среды на его развитие [1].
Задержка полового созревания является распространенным явлением и, по данным литературы, встречается у 2-3% населения, чаще у мальчиков, чем у девочек. Главным для старта полового созревания необходимо наличие нормально функционирующей системы «гипоталамус - гипофиз - гонады». Гонадотропин-рилизинг-гормон (ГнРГ), секретируемый нейронами гипоталамуса, стимулирует секрецию лютеинизирующего гормона (ЛГ) и фолликулостимулирующего гормона (ФСГ), которые, в свою очередь, способствуют созреванию половых желез и активируют выработку половых стероидов [2]. Под задержкой полового созревания у девочек понимают отсутствие или выраженное недоразвитие вторичных половых признаков в возрасте, который превышает средний статистический показатель на 2-2,5 стандартных отклонения (SD) [3]. Принято считать, что критериями задержки полового созревания у девочек являются: отсутствие вторичных половых признаков в возрасте 13 лет и старше, отсутствие менархе в возрасте 15 лет, отсутствие менархе в течение 3 лет и более от начала развития молочных желез. Для более точной оценки соответствия полового созревания возрасту разработаны нормограммы полового созревания для девочек с использованием шкалы Таннер (рис. 1) [4].
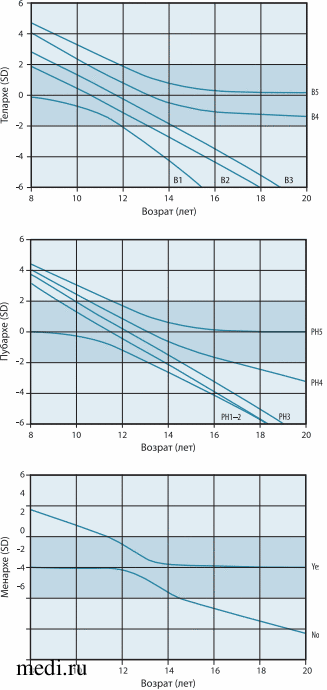 Рис. 1. Нормограмма соответствия полового созревания возрасту с использованием шкалы Таннер. Заштрихованная область показывает диапазон среднего значения ± 2 SD
Рис. 1. Нормограмма соответствия полового созревания возрасту с использованием шкалы Таннер. Заштрихованная область показывает диапазон среднего значения ± 2 SDВ зависимости от причин, приводящих к задержке полового созревания, различают перманентную форму, включающую в себя гипергонадотропный и гипогонадотропный гипогонадизм и транзиторную форму, которая представлена конституциональной задержкой полового созревания и функциональным гипогонадизмом (см. табл.) [5].
Причины задержки полового созревания у девочек
| Гипергонадотропный гипогонадизм |
|
| Гипогонадотропный гипогонадизм |
|
| Функциональный гипогонадизм |
|
Гипергонадотропный гипогонадизм является результатом первичной недостаточности яичников, что приводит к блокировке отрицательной обратной связи и повышению уровня гонадотропинов. Гипергонадотропный гипогонадизм может быть приобретенным или врожденным и в совокупности чаще встречаются у девочек, чем у мальчиков. К приобретенным причинам относят лучевую терапию для лечения рака и злокачественных новообразований, инфекции, аутоиммунное разрушение яичников. К врожденным причинам - генетические синдромы, дисгенезию гонад, дефекты стероидогенеза и другие (см. табл.) [2, 5, 6].
Гипогонадотропный гипогонадизм обусловлен нарушением функции нейронов гипоталамуса или гипофиза и характеризуется низким уровнем ЛГ и ФСГ. Состояния, вызывающие гипогонадотропный гипогонадизм, также разделяются на врожденные и приобретенные (табл.). Врожденный гипогонадотропный гипогонадизм - генетическое заболевание, возникающее в результате нарушения миграции нейронов гипоталамуса или нарушения секреции ГнРГ или инактивации специфических рецепторов гипофиза. В редких случаях причиной может являться нарушение генов, отвечающих за секрецию ЛГ и ФСГ [5-8]. И если у мальчиков врожденный гипогонадотропный гипогонадизм можно заподозрить еще в период новорожденности по таким клиническим признакам, как микропения, крипторхизм, то у девочек данное состояние начнет проявлять себя лишь в хронологическом пубертатном возрасте в виде отсутствия своевременного старта полового созревания. На сегодняшний день известно о мутациях примерно 50 генов, отвечающих за функционирование гипоталамо-гипофизарной системы [9]. Дефекты генов, влияющие на развитие передней доли гипофиза, в некоторых случаях вызывают гипопитуитаризм, включая гипогонадотропный гипогонадизм. Например, аутосомно-рецессивные мутации в гене PROP1, участвующем в формировании гипофиза, являются наиболее распространенной причиной гипопитуитаризма [10]. Причинами приобретенного гипогонадотропного гипогонадизма, в основе которого лежит нарушение синтеза или секреции ГнРГ, являются опухоли или инфекционное поражение ЦНС, травмы, лучевая/ химиотерапия, аутоиммунные заболевания [11].
Наиболее частой причиной позднего старта пубертата у обоих полов является конституциональная задержка полового созревания, и по неизвестной причине чаще встречается у мальчиков, чем у девочек (30% девочек и до 65% мальчиков) [12, 13]. По определению конституциональная задержка полового созревания является диагнозом исключения и характеризуется самостоятельным стартом полового созревания, нормальным его прогрессированием и завершением к 18 годам. Причина конституциональной задержки полового созревания неизвестна, но она имеет генетическую основу, что находит подтверждение в том, что 50-75% пациентов с конституциональной задержкой полового созревания имеют семейный анамнез задержки полового созревания, которая наследуется по аутосомно-доминантному типу с разной степенью пенетрантности [12-14].
Функциональный гипогонадизм обнаруживается примерно у 20% девочек с задержкой полового созревания или неполным половым созреванием [15, 16]. В основе патофизиологии лежит задержка созревания оси «гипоталамус - гипофиз - гонады», вторичная по отношению к хроническим заболеваниям (нервной анорексии, хроническим заболеваниям почек, воспалительным заболеваниям кишечника, серповидно-клеточной анемии и талассемии, целиакии, гипотиреозу и гиперпролактинемии, сахарному диабету, муковисцидозу), недостаточному питанию, психологическому или эмоциональному стрессу, интенсивным физическим нагрузкам (см. табл.). В этом случае воздействие на основной патогенетический механизм, включающий в себя лечение основного заболевания, оптимизацию питания, контроль физической активности восстанавливает функцию системы «гипоталамус - гипофиз -гонады» [4].
В отличие от мальчиков, функционирование женской репродуктивной системы носит циклический характер в виде менструального цикла. В связи с этим при анализе структуры задержки полового созревания у девочек необходимо учитывать такой ее нозологический вариант, как первичная аменорея, которая остается одной из трудноразрешимых проблем современной гинекологии и эндокринологии. По данным Всемирной организации здравоохранения, аменорея является шестой наиболее распространенной причиной бесплодия, а первичной аменореей страдает около 2-5% девочек-подростков [17]. Первичная аменорея может возникнуть вследствие нарушений функции гипофиза при гипогонадотропном гипогонадизме, конституциональной задержке полового созревания, функциональном гипогонадизме, заболеваниях половых желез как причине гипергонадотропного гипогонадизма, аномалиях матки и влагалища [18].
Цель исследования: изучить структуру задержки полового созревания у девочек в зависимости от этиологии и клинической картины.
Материалы и методы
В исследование включены 63 девочки в возрасте 14,92 ± 0,89 лет с задержкой полового созревания, прошедшие комплексное обследование на базе эндокринологического отделения ГБУЗ «ДГКБ им. З.А. Башляевой ДЗМ» (главный врач д-р мед. наук, профессор И.М. Османов). Критерии включения: возраст <18 лет, отсутствие вторичных половых признаков в возрасте ≥13 лет и/или отсутствие менархе в возрасте ≥15 лет, и/или отсутствие менархе в течение трех лет и более от начала появления эстрогензависимых признаков полового созревания. Критерии невключения: возраст >18 лет, неправильное строение наружных половых органов.
Всем пациенткам оценивали стадию полового созревания по шкале Таннер, антропометрические показатели, костный возраст по методу Грейлиха -Пайля, генитометрические показатели (данные УЗИ органов малого таза трансабдоминальным доступом), содержание лютеинизирующего гормона (ЛГ; 1,9-14,2 мМЕ/мл), фолликулостимулирующего гормона (ФСГ; 2,5-11,2 мМЕ/мл), эстрадиола (Э2; 30393 пмоль/л), ингибина В (20-177 пг/мл, n = 50), антимюллерова гормона (АМГ; <8,9 нг/мл, n = 55), пролактина (70-600 мМЕ/л), тестостерона (0,291,67 нмоль/л), инсулиноподобного фактора роста -1 (ИФР-1; 188,4-509,9 нг/мл), тиреотропного гормона (ТТГ; 0,465-4,680 мМЕ/мл), свободного Т4 (7,8614,41 пмоль/л) в сыворотке крови, результаты теста с аналогом ГнРГ (трипторелин 0,1 мг, n = 39). Показатели оценивали по нормативам девочек. Проводили цитогенетическое исследование (n = 24), молекулярно-генетический анализ методом высокопроизводительного параллельного секвенирования (n = 33, панель custom Ampliseq_DSD (нарушения формирования пола): гены AKR1C2, AKR1C4, AMH, AMHR2, AR, ARX, ATRX, CBX2, CYB5A, CYP11A1, CYP17A1, DHCR7, DHH, EMX2, ESR2, FGD1, FGF9, FGFR2, FKBP4, FOXF2, FOXL2, HOXA13, HSD1733, HSD382, ICK, LHCGR, LHX1, LHX9, MAMLD1, MAP3K1, MID1, NROB1, NR5A1, POR, PTGDS, SOX9, SRD5A2, SRY, STAR, SUPT3H, TSPYL1, WNT4, WT1, ZFPM2 и панель custom Ampliseq_HP (гипопитуитаризм): гены FGF8, NELF, SEMA3A, KISS1, KAL2, KAL1, TACR3, WDR11, GREAT, GNRH1, TAC3, KAL4, NROB1, LHB, PROKR2, GNRHR, KISSIR, CHD7, HSEST1, INSL3, IL17RD, SPRY4, FGF17, DUSP6, FLRT3, DNMTBL, POLRA3A, POLR3B, RBM28, MKRN3, около 106 000 пар оснований, общее покрытие 96,6%), собирали сведения о соматических заболеваниях, изучали наследственность по задержке полового созревания в семье.
Согласно дизайну исследования, каждые 6 месяцев проводился телефонный контакт для оценки темпов полового созревания. Законными представителями пациенток было подписано информированное согласие на обследование и обработку полученных результатов.
Статистический анализ проводился с использованием программы StatTech v. 4.2.7 (разработчик -ООО «Статтех», Россия). Категориальные данные описывались с указанием абсолютных значений и процентных долей. Сравнение процентных долей при анализе многопольных таблиц сопряженности выполнялось с помощью критерия хи-квадрат Пирсона, различия считались статистически значимыми при p < 0,05.
Пациенты были отнесены к группе гипергонадотропного гипогонадизма на основании высокого содержания базальной концентрации гонадотропных гормонов в сыворотке крови, диагноз уточнялся путем выполнения цитогенетического исследования и молекулярно-генетического анализа. К группе гипогонадотропного гипогонадизма относили девочек с низкой базальной концентрацией гонадотропинов в сыворотке крови, уровнем ингибина В <20 пг/мл, максимальным подъемом ЛГ по данным теста с аналогом ГнРГ <10 мМЕ/мл, или с подтвержденной патологией ЦНС / генетическим дефектом, а также при отсутствии спонтанного прогрессирования полового созревания в течение периода наблюдения до возраста 18 лет. В качестве диагностических критериев конституциональной задержки полового созревания считали максимальный подъем ЛГ по данным теста с аналогом ГнРГ > 10 мМЕ/мл, отягощенный анамнез по наличию позднего пубертата в семье, а также отсутствие клинических, биохимических и визуализирующих данных, которые могли указывать на патологические причины, лежащие в основе задержки полового созревания. Диагноз функциональный гипогонадизм устанавливался в том случае, если у пациенток было выявлено основное заболевание, которое, вероятно, задерживало половое созревание, при этом отмечалось спонтанное прогрессирование вторичных половых признаков во время наблюдения, при отсутствии лабораторных данных, указывающих на наличие перманентной формы задержки полового созревания.
Результаты исследования
Структура задержки полового созревания в зависимости от этиологической причины представлена в 38% (24/63) случаев гипергонадотропным гипогонадизмом, в 25,4% (16/63) - конституциональной задержкой полового созревания, в 20,6% (13/63) случаев функциональным гипогонадизмом и в 16% (10/63) -гипогонадотропным гипогонадизмом (рис. 2). В целом группы перманентной и транзиторной форм сопоставимы по количеству пациенток (54% (34/63) vs 46% (29/63 соответственно, p = 0,380).
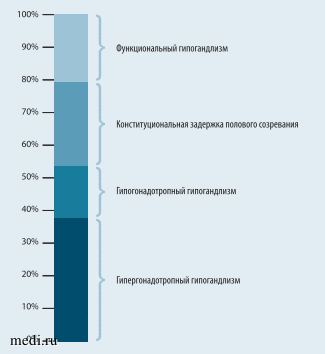 Рис. 2. Структура задержки полового созревания (n = 63)
Рис. 2. Структура задержки полового созревания (n = 63)
По результатам цитогенетического анализа 45,8% (11/24) пациенток с гипергонадотропным гипогонадизмом имели вариации кариотипа, соответствующего диагнозу синдром Шерешевского - Тернера, 29,2% (7/24) пациенток имели кариотип 46, XY, 25% (6/24) - кариотип 46, XX.
Среди девочек с моносомией по Х-хромосоме по результатам цитогенетического анализа у 27% (3/11) выявлен кариотип 45, X/46, XX, что соответствует мозаичному варианту синдрома Шерешевского -Тернера.
Молекулярно-генетический анализ группы девочек с мужским кариотипом выявил у двух пациенток с отсутствием дериватов мюллерова протока - полную нечувствительность к андрогенам, обусловленную гемизиготной мутацией гена AR. В оставшихся четырех случаях установлена тотальная дисгенезия гонад, в двух случаях SRY негативный вариант, в двух случаях SRY позитивный, в одном случае - обусловленный гетерозиготной мутацией гена WT1.
В группе девочек с кариотипом 46, XX точные причины гипергонадотропного гипогонадизма не установлены.
Гипогонадотропный гипогонадизм в 20% (2/10) случаев входил в состав гипопитуитаризма (в одном наблюдении носил приобретенный характер - после удаления краниофарингиомы, во втором - врожденный, вследствие дефекта гена PROP1. В остальных 80% (8/10) случаев имела место изолированная форма заболевания, у 50% (4/8) девочек диагноз подтвержден выявлением патогенных мутаций в гене GNR: в одном случае выявлены гетерозиготные варианты с.21Т>С:р.М1Т и c.416G>A:p.R139H; во втором случае выявлен гомозиготный вариант c.416G>A:p.R139H; в третьем случае обнаружена компаунд-гетерозиготная мутация; в четвертом случаев в 1 экзоне обнаружен вариант нуклеотидной последовательности в гетерозиготном состоянии, ранее описанный как патогенный.
Анализ структуры функционального гипогонадизма показал, что задержку полового созревания вследствие декомпенсации сахарного диабета типа 1 имели 46,2% (6/13), в результате белково-энергетической недостаточности - 46,2% (6/13), целиакии -7,6 % (1/13) пациенток.
При изучении структуры задержки полового созревания у девочек в зависимости от клинических проявлений в группе гипергонадотропного гипогонадизма причинами обращения было отсутствие вторичных половых признаков у 70,8% (17/24) пациенток, первичная аменорея у 29,2% (7/24).
В группе гипогонадотропного гипогонадизма отсутствие вторичных половых признаков наблюдалось у 60% (6/10) пациенток, первичная аменорея у 40% (4/10).
Среди девочек с конституциональной задержкой полового созревания отсутствие вторичных половых признаков имели 31,3% (5/16) пациенток, первичную аменорею 68,7% (11/16) пациенток.
В группе функционального гипогонадизма причинами обращения было отсутствие вторичных половых признаков у 38,5% (5/13) пациенток, первичная аменорея у 61,5% (8/13) пациенток.
При сравнении перманентной и транзиторной форм задержки полового созревания в структуре перманентной формы значимо чаще пациентки обращались с жалобами на отсутствие вторичных половых признаков, тогда как в группе с транзиторной формой задержки полового созревания основной причиной обращения была первичная аменорея (68% (23/34) vs 66% (19/29) соответственно, р = 0,009).
Обсуждение
По результатам проведенного исследования ведущей причиной задержки полового созревания являлся гипергонадотропный гипогонадизм. Однако, по данным литературы, в структуре задержки полового созревания у девочек преобладает конституциональная задержка полового созревания или функциональный гипогонадизм [12, 13, 19-21].
В свою очередь, среди причин гипергонадотропного гипогонадизма лидировал синдром Шерешевского - Тернера. Значительно реже встречалось нарушение формирование пола 46, ХҮ, представленное в большей степени тотальной дисгенезией гонад и нарушением формирования пола 46, ХХ, что соответствует частоте распространения данных состояний в популяции [22].
Второй по частоте причиной задержки полового созревания, по результатам исследования, являлась конституциональная задержка полового созревания, на ее долю приходилось более четверти всех проанализированных случаев.
На третьем месте - функциональный гипогонадизм, обусловленный одинаково часто хронической декомпенсацией сахарного диабета тип 1 и белково-энергетической недостаточностью.
Наиболее редким патологическим состоянием, приводящим к задержке полового созревания, являлся гипогонадотропный гипогонадизм, установленный в 16% всех случаев, что также соответствует данным литературы [5]. В структуре гипогонадотропного гипогонадизма лишь один случай приобретенной формы, вследствие удаления краниофарингиомы, в остальных наблюдениях имела место врожденная форма, которая в половине случаев генетически подтверждена.
Если говорить о причинах обращения, то жалобы на отсутствие вторичных половых признаков чаще предъявляли девочки с гипогонадотропным и гипергонадотропным гипогонадизмом, тогда как с жалобами на первичную аменорею чаще обращались девочки с транзиторной формой задержки полового созревания.
Выводы
Задержка полового созревания является частой причиной обращения к детскому эндокринологу. Знание структуры задержки полового созревания помогает понять причину данного состояния и выбрать правильную траекторию диагностического поиска, что способствует более быстрой постановке диагноза и оптимальной тактике ведения.
Результаты проведенного исследования показали, что структура задержки полового созревания у девочек в равных долях представлена транзиторной и перманентной формами. Ведущей этиологической причиной задержки полового созревания является гипергонадотропный гипогонадизм, наиболее редкой - гипогонадотропный гипогонадизм. Среди пациенток с гипергонадотропным гипогонадизмом у каждой второй девочки диагностирован синдром Шерешевского - Тернера, у каждой третьей - выявлен мужской кариотип, что чаще всего соответствовало диагнозу «чистая дисгенезия гонад 46, XY», а каждая четвертая имела нормальный женский кариотип. Среди пациенток с гипогонадотропным гипогонадизмом изолированная форма встречалась наиболее часто и в равных долях подтверждение диагноза было представлено генетической мутацией и данными динамического наблюдения. Функциональный гипогонадизм в равной степени представлен декомпенсацией сахарного диабета тип 1 и белково-энергетической недостаточностью, и лишь в одном случае целиакией. Клиническая картина заболевания, характеризующаяся отсутствием вторичных половых признаков, чаще встречалась в структуре перманентной формы задержки полового созревания, а в случаях первичной аменореи -в структуре транзиторной формы.
Литература
1. Charles Sultan MD, PhD, Pr, Laura Gaspari MD, Laurent Maimoun PhD , Nicolas Kalfa MD, PhD, Pr , Francoise Paris MD, PhD, Pr Disorders of puberty. Best Practice & Research Clinical Obstetrics & Gynaecology Volume 48, April 2018, Pages 62-89
2. Palmert MR & Dunkel L. Clinical practice. Delayed puberty // New England Journal of Medicine. 2012. Vol. 366. P. 443-453. DOI:10.1056/ NEJMcp1109290.
3. Raivio Т., Miettinen P.J. Constitutional delay of puberty versus congenital hypogonadotropic hypogonadism: Genetics, management and updates // Best Practice 2 & Research Clinical Endocrinology & Metabolism . 2019. Vol. 33. P. 101316/
4. Pubertal Progression and Reproductive Hormones in Healthy Girls With Transient Thelarche / M.L. Johansen [et al.] // The Journal of Clinical Endocrinology & Metabolism. 2017. Vol. 102, Issue 3, 1. P 1001-1008. DOI: org/10.1210/jc.2016-2871.
5. Ines L. Sedlmeyer, Mark R. Palmert, Delayed Puberty: Analysis of a Large Case Series from an Academic Center // The Journal of Clinical Endocrinology & Metabolism. 2002. Vol. 87. Issue 4, 1. P 1613-1620. DOI: org/10.1210/jcem.87.4.8395.
6. Tang C., Zafar Gondal A., Damian M. Delayed Puberty. [Updated 2023 Jul 31]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https:// www.ncbi.nlm.nih.gov/books/NBK544322.
7. Expert consensus document: European Consensus Statement on congenital hypogonadotropic hypogonadism: pathogenesis, diagnosis and treatment / U. Boehm [et al.] // Nat Rev Endocrinol. 2015. Vol. 11. P. 547-564.
8. Clinical management of congenital hypogonadotropic hypogonadism / J. Young [et al.] // Endocrine Reviews. 2019. Vol. 40. P 669-710.
9. Topaloglu A.K. Update on the Genetics of Idiopathic Hypogonadotropic Hypogonadism // Journal of Clinical Research in Pediatric Endocrinology. 2017. Dec. 30. Vol. 9 (Suppl 2). P 113-122. DOI: 10.4274/jcrpe. 2017.S010. Epub 2017 Dec 27. PMID: 29280744; PMCID: PMC5790323.
10. Heritable disorders ofpituitary development / J.S. Parks [et al.] // The Journal of Clinical Endocrinology & Metabolism. 1999. Vol. 84, No. 12. P 4362-4370.
11. Howard S.R., Dunkel L. Delayed puberty - phenotypic diversity, molecular genetic mechanisms, and recent discoveries // Endocrine Reviews. 2019. Vol. 40. P. 1285-1317.
12. Sedlmeyer I.L., Hirschhorn J.N., Palmert M.R. Pedigree analysis of constitutional delay of growth and maturation: determination of familial aggregation and inheritance patterns // The Journal of Clinical Endocrinology & Metabolism. 2002. Vol. 87. P 5581-5586.
13. Patterns of inheritance of constitutional delay of growth and puberty in families of adolescent girls and boys referred to specialist pediatric care / K. Wehkalampi [et al.] // The Journal of Clinical Endocrinology & Metabolism. 2008. Vol. 93. P. 723-728.
14. Genetic determinants of pubertal timing in the general population / Z.K. Gajdos, K.D. Henderson, J.N. Hirschhorn, M.R. Palmert // Molecular and Cellural Endocrinology. 2010. Vol. 324. P 21-29.
15. Howard S.R., Dunkel L. Delayed puberty - phenotypic diversity, molecular genetic mechanisms, and recent discoveries // Endocrine Reviews. 2019. Vol. 40. P. 1285-1317.
16. Congenital hypogonadotropic hypogonadism, functional hypogonadotropism or constitutional delay of growth and puberty? An analysis of a large patient series from a single tertiary center / T. Varimo [et al.] // Human Reproduction. 2017. Vol. 32. P 147-153.
17. Primary Amenorrhea with hypothyroidism: Finding the Cause / A.M. Alvi [et al.] // Cureus. 2021. Dec. 8. Vol. 13, No. 12. P e20285. DOI: 10.7759/cureus.20285. PMID: 35018272; PMCID: PMC8742137.
18. Management of endocrine disease: diagnosis and management of primary amenorrhea and female delayed puberty / S. Seppa, T. Kuiri-Hanninen, E. Holopainen, R. Voutilainen // European Journal of Endocrinology. 2021. Vol. 184. P. 0-42.
19. Timing of Pubertal Onset in Girls and Boys With Constitutional Delay / E. Jonsdottir-Lewis [et al.] // The Journal of Clinical Endocrinology & Metabolism. 2021. Vol. 106. Issue 9. P e3693-e3703. DOI: org/10.1210/clinem/dgab270.
20. Palmert, Pedigree Analysis of Constitutional Delay of Growth and Maturation: Determination of Familial Aggregation and Inheritance Patterns / L. Ines [et al.] // The Journal of Clinical Endocrinology & Metabolism. 2002. Vol. 87. Issue 12, 1 December. P 5581-5586. DOI: org/10.1210/jc.2002-020862.
21. Tang C., Zafar Gondal A., Damian M. Delayed Puberty. [Updated 2023 Jul 31]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan. Available from: https:// www.ncbi.nlm.nih.gov/books/NBK544322.
22. Bondy C.A. Turner Syndrome Study Group. Care of girls and women with Turner syndrome: a guideline of the Turner Syndrome Study Group // The Journal of Clinical Endocrinology & Metabolism. 2007. Vol. 92, No. 1. P. 10-25.
Комментарии
ПРАКТИКА ПЕДИАТРА



