Особенности клинической картины и диагностики синдрома Бругада у детей и подростков: обзор литературы
СтатьиА.П. Смирнова, Б.А. Павликов, А.В. Еремеева, канд. мед. наук,
ФГБОУ ВО «Первый Московский государственный медицинский университет им. И.М. Сеченова» Минздрава России
Резюме. Внезапная сердечная смерть у детей встречается с частотой 0,7-6,4 случая на 100 тыс. Ведущей причиной ее возникновения считаются злокачественные аритмии. Одним из состояний, приводящих к их развитию, является синдром Бругада. Синдром Бругада - генетически и фенотипически гетерогенное наследственное заболевание, характеризующееся злокачественным клинико-электрографическим аритмическим синдромом, который проявляется в виде элевации сегмента ST в правых грудных отведениях (V1-V3) сводчатой конфигурации (так называемый Бругада-паттерн I типа), блокадой правой ножки пучка Гиса на электрокардиограмме (ЭКГ) и рецидивирующими обмороками или эпизодами внезапной сердечной смерти вследствие полиморфной желудочковой тахикардии или фибрилляции желудочков. Несмотря на то, что на сегодняшний день был достигнут заметный прогресс в понимании патофизиологических процессов заболевания и предложены критерии диагностики и стратификации риска для взрослых пациентов, у детей диагностика данного заболевания затруднена в связи с высокой частотой бессимптомного течения и несовершенством интерпретации записи ЭКГ. Согласно ряду исследований, самые частые проявления данного синдрома у детей - случайные ЭКГ-находки, а синкопальные состояния и аритмии в данном возрасте регистрируются значительно реже, также важно изучение семейного анамнеза. ЭКГ во время действия провоцирующих электрокардиографические изменения факторов, таких как фебрильная температура, прием лекарств и высокий парасимпатический тонус, обусловливает большие возможности для диагностики данного синдрома у пациентов инфекционных отделений при условии записи ЭКГ в период лихорадки и у пациентов, которым проводилось суточное мониторирование ЭКГ по Холтеру. Также важным аспектом в стратификации риска у детей является проведение генетического исследования, так как была выявлена более высокая, чем у взрослых, частота развития злокачественных аритмий и их рецидивов у пациентов, имеющих подтвержденную мутацию в гене SCN5A.
Ключевые слова: синдром Бругада, внезапная сердечная смерть, диагностика, каналопатия
Для цитирования: Смирнова А.П., Павликов Б.А., Еремеева А.В. Особенности клинической картины и диагностики синдрома Бругада у детей и подростков: обзор литературы // Практика педиатра. 2021. № 1. С. 69-76.
Summary. Sudden cardiac death (SCD) among children is 0,7-6,4 cases per 100 000 population. The leading mechanism of SCD in this case is malignant arrhythmias. One of the conditions leading to their development is Brugada syndrome. Brugada syndrome is a genetically and phenotypically heterogeneous hereditary disease characterized by malignant clinical and electrographic arrhythmic syndrome, which manifests itself in the form of ST segment elevation in the right thoracic leads (V1 - V3) of a vaulted configuration (the socalled Brugada pattern type I), blockade of the right leg of the Gis bundle on an ECG and clinically in the form of recurrent syncope or episodes of sudden cardiac death due to polymorphic ventricular tachycardia or ventricular fibrillation. Despite the fact that significant progress has been made to date in understanding the pathophysiological processes of the disease and criteria for diagnosis and risk stratification for adult patients have been proposed, diagnosis of this disease in children is difficult due to the high frequency of asymptomatic course and imperfect interpretation of the electrocardiogram recording. According to a number of studies, the most common manifestations of this syndrome in children are a hereditary history and accidental ECG findings, syncopal conditions and arrhythmias at this age are recorded much less frequently. ECG recording can expand diagnostic capabilities during the action of factors provoking electrocardiographic changes, such as febrile temperature, medications and periods of high parasympathetic tone, which leads to great opportunities for diagnosing this syndrome in patients of infectious departments, provided that an ECG is recorded during fever and in patients who underwent daily ECG monitoring by Holter. Another important aspect in the stratification of risk for children is the conduct of a genetic study, since a higher incidence of malignant arrhythmias and their recurrence among patients with a confirmed mutation in the SCN5A gene was revealed than in adults.
Keywords: Brugada syndrome, sudden cardiac death, diagnosis, channelopathy
For citation: Smirnova A.P., Pavlikov B.A., Eremeeva A.V. Features of the clinical picture and diagnosis of Brugada syndrome in children and adolescents: literature review // Pediatrician's Practice. 2024;(1):69-76. (In Russ.)
Введение
Внезапная сердечная смерть (ВСС) составляет подавляющее большинство случаев внезапной смерти. Согласно данным многочисленных международных исследований, ВСС у детей встречается с частотой 0,7-6,4 случая на 100 тыс. Ведущим механизмом ее возникновения считаются злокачественные аритмии [1]. Следовательно, путь к первичной профилактике ВСС - своевременная диагностика наследственных аритмий и каналопатий, приводящих к жизнеугрожающим аритмиям. Одним из таких состояний является синдром Бругада (СБ). Несмотря на то, что данный синдром встречается в детской популяции значительно реже (0,0098%), чем среди взрослых (от 0,1 до 0,5%), именно СБ является причиной 4-12% всех случаев ВСС [2].
СБ - генетически и фенотипически гетерогенное наследственное заболевание, характеризующееся злокачественным клинико-электрографическим аритмическим синдромом, который проявляется спонтанной или спровоцированной персистирующей или рецидивирующей элевацией сегмента ST сводчатой конфигурации в правых грудных отведениях (Бругада-паттерн I типа), блокадой правой ножки пучка Гиса на ЭКГ и рецидивирующими обмороками или эпизодами ВСС вследствие злокачественных желудочковых аритмий (полиморфной желудочковой тахикардии или фибрилляции желудочков). Впервые СБ описан братьями Педро и Жозепом Бругада в 1992 г., когда они доложили о 8 успешно реанимированных пациентах с фибрилляцией желудочков, у которых наблюдался характерный клинико-электрокардиографический симптомокомплекс [2].
С тех пор был достигнут заметный прогресс в понимании основных механизмов развития заболевания, стратификации риска ВСС и желудочковых аритмий у пациентов с СБ. С клинической точки зрения, хотя электрокардиографические критерии СБ широко описаны в научной литературе, его диагностика, особенно у детей, остается непростой задачей в связи с бессимптомным течением и несовершенством провокационных проб, а также с риском жизнеугрожающих аритмий, сопряженных с ними, и сложностью определения критериев риска у бессимптомных пациентов или пациентов с Бругада-паттернами II и III типов. Также в последние годы все чаще обсуждается наличие у пациентов с СБ структурных аномалий сердца [1, 2].
Эпидемиология
Наиболее распространен СБ в Юго-Восточной Азии - 15 случаев на 10 тыс. человек, а самая высокая частота СБ наблюдается в Таиланде - 6,8 на 1000 человек, что примерно в 14 раз выше общемировой частоты встречаемости [3].
Согласно результатам исследования A. Milman и соавт., среди детей с генетически подтвержденным СБ преобладают девочки, в то время как среди пациентов с СБ в возрасте от 16 до 70 лет преобладают мужчины [4]. Аналогично в исследовании D. Righi и соавт. наблюдалась более высокая частота злокачественных желудочковых аритмий и обмороков у пациентов женского пола, хотя различия не были статистически значимыми из-за небольшого размера выборки. Дальнейшее снижение склонности к аритмиям у женщин в постпубертатном периоде по сравнению с показателем пациентов мужского пола может быть связано с действием женских половых гормонов, которые уменьшают экспрессию каналов Ito (транзиторный калиевый ток) в эпикарде правого желудочка [5]. Однако для подтверждения этих данных необходимы дальнейшие исследования с более крупными когортами пациентов.
Патофизиология
На данный момент существуют три гипотезы, объясняющие патофизиологические механизмы развития синдрома Бругада: гипотеза реполяризации, гипотеза деполяризации и гипотеза нервного гребня.
Гипотеза реполяризации основывается на возникновении трансмурального градиента напряжения между субэпикардиальным и субэндокардиальным слоями правого желудочка, что приводит к развитию летальных аритмий. Уменьшение суммарного натриевого тока (INa) и повышении внешнего калиевого тока (Ito) вызывает разницу зарядов в эпикарде правого желудочка, происходит преждевременная реполяризация кардиомиоцитов. Таким образом, короткий потенциал действия относительно эндокарда приводит к образованию трансмурального градиента напряжения, который электрокардиографически проявляется в виде характерного подъема сегмента ST. Развитию второй фазы re-entry способствует эпикардиальная дисперсия реполяризации, возникающая в результате диасбаланса токов и потери купола потенциала действия. Это приводит к экстрасистоле, провоцирующей желудочковые аритмии [2, 3].
Согласно гипотезе деполяризации, характерный подъем сегмента ST по типу «свода» на ЭКГ возникает вследствие разности потенциалов между участками правого желудочка и выводным трактом правого желудочка (ВТПЖ). Причиной разности потенциалов может быть задержка проводимости в ВТПЖ, а желудочковые аритмии при СБ индуцируются аномальным током, возникающим в результате замедленной деполяризации ВТПЖ [2]. В исследовании K. Nademanee и соавт. по оценке ВТПЖ у людей с СБ и без него (посмертно или in vivo во время эпикардиальной абляции по поводу аритмии с помощью торакотомии) у пациентов с СБ были обнаружены гистологические признаки эпикардиального поверхностного и внутримиокардиального фиброза, а также снижение экспрессии белка щелевого контакта - коннексина-43 [6]. Аналогично в исследовании D. Corrado и соавт. у 12 из 13 пациентов, умерших от ВСС с ЭКГ-картиной СБ, обнаружены участки фиброза субэпикардиального миокарда правого желудочка [7]. Таким образом, данные исследования подтверждают большое значение аномальной структуры миокарда (увеличение коллагена, эпикардиальный и интерстициальный фиброз, снижение экспрессии белка щелевого контакта) в патофизиологии заболевания, несмотря на отсутствие явных структурных заболеваний сердца у пациентов с СБ.
Гипотеза нервного гребня была предложена Elizari и соавт. Так как клетки нервного гребня играют важную роль в развитии миокарда ВТПЖ и соседних структур, можно предположить, что нарушение их экспрессии может приводить к аномальной миокардиализации. Ремоделирование ВТПЖ, в свою очередь, ведет к неравномерному распределению коннексина, особенно коннексина-43, как в толще миокарда, так и в различных отделах сердца. Замедление деполяризации участков, обедненных коннексином, и разница потенциалов между участками миокарда обусловливают гетерогенную реполяризацию ВТПЖ при СБ [2].
Генетические аспекты
СБ - генетически обусловленное заболевание, наследуемое по аутосомно-доминантному типу. Около 30% пациентов имеют мутацию в гене SCN5A на коротком плече 3-й хромосомы (3p21-24). Существует ряд других генов, мутация которых может привести к СБ (SCN5A, GPD1L, SCN1B, SCN2B, SCN3B, RANGRF, SLMAP, KCNE3, KCNJ8, HCN4, KCNE5, KCND3, CACNA1C, CACNB2B, CACNA2D1 и TRPM4), однако мутации в них выявляют лишь в 5% случаев [8]. Они кодируют субъединицы сердечных натриевых, калиевых и кальциевых каналов и приводят к нарушению входящего натриевого или кальциевого тока либо увеличению выходящего калиевого тока. У 65% пациентов мутация остается недиагностированной.
Ген SCN5A кодирует синтез сердечной а-субъединицы потенциал-зависимого натриевого канала Nav1.5, через который осуществляется быстрый деполяризующий натриевый ток (INa) в первую фазу потенциала действия и поздний входящий натриевый ток (INaL), влияющий на реполяризацию и рефрактерность. Мутации в данном гене снижают пиковый компонент INa, ведут к дисфункции канала и проявляются рядом фенотипов, включая СБ, синдром слабости синусного узла и болезнь Ленегра - Лева. Мутации гена SCN5A также могут обусловливать сложный фенотип заболевания, включающий не только СБ, но и брадикардию, дефекты проводимости, удлиненный интервал QT. Мутация 1795insD приводит к синдрому слабости синусового узла, нарушениям проводимости, СБ и удлиненному интервалу QT. Такое генетически обусловленное развитие сразу нескольких нарушений проводимости, связанных с одним геном, описывается в литературе как перекрестный (overlap) синдром. При этом возникает клиническое и генетическое перекрытие между аритмическими синдромами, ранее считавшимися самостоятельными заболеваниями. Механизмы развития перекрестного синдрома до конца не изучены. Провести границу между такими разными электрофизиологическими сущностями сложно, но необходимо, так как это позволяет принять решение о необходимой терапии [9]. С другой стороны, наличие у пациента одного из данных синдромов обусловливает необходимость более тщательной диагностики других фенотипических проявлений данной мутации для исключения или подтверждения перекрестного синдрома.
Предполагают, что полиморфизм митохондриальной ДНК (мтДНК) также может быть причиной вариабельности фенотипа заболевания, так как дисфункция митохондрий, в том числе связанная с различными вариантами мтДНК, может оказывать аритмогенный эффект за счет уменьшения отношения АТФ/АДФ, открытия калиевых каналов, замедления проведения импульса. Кроме того, снижается поступление в клетку натрия и кальция и, в результате, возбудимость кардиомиоцитов. Таким образом, дисфункция мтДНК вызывает изменения, схожие с эффектом мутаций при СБ - снижение пикового компонента INa и раннюю реполяризацию. Возникающие мутации мтДНК образуют устойчивые гаплотипы, их объединяют в гаплогруппы. В итальянской популяции, по данным L. Stocchi и соавт., гаплогруппы T и J, объединяемые общим полиморфизмом T4216C, встречаются у всех пациентов со спонтанным Бругада-паттерном I типа на ЭКГ и характерной клинической картиной. В результате исследования было предположено, что принадлежность мтДНК к гаплогруппам T и J является важным модифицирующим фактором, создающим условия для проявления фенотипа СБ [10]. Опровержение этой ассоциации можно найти в исследовании М.В. Голубенко и соавт. В российской популяции суммарная частота гаплогрупп J и Т также составляет около 20%, однако в ходе исследования у больных СБ была обнаружена только гаплогруппа Т - 11% [11].
Генетическое исследование является важным для оценки риска развития жизнеугрожающих аритмий у детей и подростков. Анализ данных выявил значительно большую распространенность злокачественных аритмий у пациентов с мутациями SCN5A по сравнению с другими пациентами. D. Righi и соавт. описывают значительную связь между мутациями SCN5A и возникновением злокачественных аритмий у детей младше 12 лет [5]. Также, согласно данным Y. Michowitz и соавт., у подростков с мутациями SCN5A отмечается повышенный риск повторных аритмических событий [12].
Клиническая картина
Выраженность клинических проявлений при СБ зависит от степени нарушения функции натриевых каналов: при дисфункции менее 25% каналов отмечается бессимптомное течение, ЭКГ-изменения и клиническая картина наблюдаются после провокации блокаторами натриевых каналов. При повреждении более 25% натриевых каналов риск ВСС значительно возрастает, возникает спонтанный Бругада-паттерн I типа, синкопальные состояния [8].
В большинстве случаев СБ у детей течет бессимптомно (72%), однако у ряда пациентов первым проявлением заболевания может быть ВСС вследствие желудочковых аритмий. В отличие от пациентов с синдромом удлиненного интервала Q-T, которые часто имеют предупреждающие симптомы в виде рецидивирующего синкопе, прежде чем у них произойдет остановка сердца, у пациентов с СБ часто первые же аритмические события приводят к летальному исходу [13]. Согласно исследованиям, очень часто одним из проявлений СБ у детей является отягощенный семейный анамнез (47%), затем случайные ЭКГ-находки (25%), обмороки (14%), аритмии (13%), самым редким проявлением - прерванная ВСС (1%) [2].
Большинство синкопальных эпизодов возникают в покое ночью и часто провоцируются лихорадкой (45%), которая, в свою очередь, может быть вызвана в том числе вакцинацией [2]. Доказана связь лихорадки не только с развитием синкопе, но и с реализацией наследственных аритмий. Первое сообщение о возникновении фибрилляции желудочков на фоне фебрильной температуры тела у больного с СБ было опубликовано в 2000 г. Показано ее влияние на потенциал-зависимые калиевые каналы: происходит снижение тока калия, что приводит к удлинению интервала QT, особенно в условиях гипокалиемии. У детей рецидивы аритмий также часто связаны с лихорадкой [1]. Элевация сегмента ST связана и с высокой парасимпатической активностью, поэтому часто клинический проявления СБ наблюдаются ночью или во время еды и могут быть спровоцированы приемом вагомиметиков.
Также увеличение высоты сегмента ST ночью связано с повышением уровня тестостерона в сыворотке крови. В исследовании W. Shimizu и соавт. была выявлена прямая зависимость между частотой аритмических событий и уровнем тестостерона крови у мужчин и обратная зависимость между частотой аритмический событий и индексом массы тела [14]. Алкоголь также может спровоцировать приступ желудочковой аритмии у пациентов с СБ.
Диагностика
Выделяют 3 типа электрокардиографических паттернов СБ.
Паттерн I типа характеризуется подъемом сегмента ST ≥ 2 мм по сводчатому типу (соуеd type) с последующим отрицательным Т-зубцом. Иногда данный тип элевации сегмента ST называется конфигурацией «бультерьера». Наличие сводчатой конфигурации сегмента ST преобладает при симптомных формах СБ.
Паттерн II типа также характеризуется элевацией ST, однако она имеет седловидную форму (saddleback type). Зубец T при этом положительный или двухфазный. Данный тип электрокардиографических изменений чаще встречается при бессимптомных формах СБ.
Паттерн III типа характеризует элевация сегмента ST < 1 мм, которая может быть обеих конфигураций, но чаще отмечают «седловидную» (см. рисунок). Пациенты с данным типом паттерна также чаще бессимптомны.
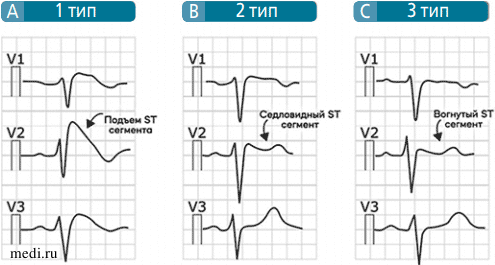 Электрокардиографические паттерны синдрома Бругада.
Электрокардиографические паттерны синдрома Бругада. Высокие правые грудные отведения наиболее информативны для исследования области ВТПЖ, поэтому диагностическая значимость ЭКГ при СБ повышается при расположении электродов в правых грудных отведениях на 1-2 межреберья выше. Следует также помнить, что ЭКГ-проявления СБ уменьшаются или исчезают при пробе с физической нагрузкой и при лекарственной пробе с симпатомиметиками.
Диагностическим критерием СБ, согласно предложенным в 2013 г. рекомендациям HRS/EHRA/ APHRS, считается наличие спонтанного или лекарственно-индуцированного ЭКГ-паттерна I типа, зарегистрированного в одном или нескольких правых грудных отведениях (V1, V2) [15].
Таким образом, диагностика СБ подразумевает использование фармакологических и нагрузочных тестов. Для диагностики СБ чаще используют фармакологическую нагрузочную пробу с антиаритмическими препаратами IA класса - блокаторами натриевых каналов (новокаинамид, аймалин). Лекарственная проба может вызывать подъем сегмента ST при раннем отсутствии изменений или усиливать уже имеющуюся элевацию с изменением типа паттерна. При этом диагностической считается модификация II и III типа ЭКГ-паттернов в паттерн I типа, а также достижение J-волной амплитуды >2 мм в правых грудных отведениях. Проба сомнительна при изменении III типа ЭКГ на II тип. В таком случае необходимо проводить дополнительные диагностические исследования для определения диагноза.
При рутинном применении провокационного теста с аймалином и оценке ЭКГ-изменений в правых прекардиальных отведениях СБ оказался наиболее распространенным диагнозом среди наследственных заболеваний сердца, выявленных в семьях с ВСС [16]. Высокая чувствительность теста с аймалином для обнаружения СБ предполагает, что пациенты с отрицательным результатом теста имеют благоприятный прогноз, несмотря на то что доказательств этого утверждения крайне мало. Исследование Brugada и соавт. продемонстрировало высокую чувствительность аймалинового теста и меньшую чувствительность флекаинида и прокаинамида в диагностике СБ. В рамках исследования ни у одного из 53 пациентов контрольной группы при проведении провокационного теста не развился ЭКГ паттерн I типа. Однако данное исследование не позволяет сделать выводы о специфичности теста из-за того, что у 8 пациентов контрольной группы была дисплазия правого желудочка. Дальнейшие исследования с большим числом пациентов продемонстрировали 16% ложноположительных результатов аймалиновой пробы при дисплазии правого желудочка. Также у 10 человек из контрольной группы была полная блокада правой ножки пучка Гиса, что может маскироваться под ЭКГ-паттерн I типа. Отсутствие ложноположительных результатов в контрольной группе, возможно, обусловлено тем, что ни у кого из контрольной группы электроды для регистрации ЭКГ не были размещены в третьем или втором межреберье, что используется в общей практике для регистрации СБ, а использование высоких электродов почти удваивает чувствительность теста и снижает специфичность. Таким образом, проведение провокационного теста с аймалином должно быть ограничено неопределенной специфичностью теста и значительным проаритмическим риском (с провокацией фибрилляции желудочков максимально у 1,8% обследованных пациентов) [17].
Для исключения гипердиагностики СБ у пациентов с лекарственно индуцированным Бругада-паттерном I типа была предложена шанхайская шкала для диагностики СБ - новые диагностические критерии, которые учитывают результаты лекарственно спровоцированных изменений на ЭКГ лишь в сочетании с клиническими проявлениями, семейным анамнезом и результатами генетического тестирования. Таким образом, отличие шанхайской диагностической шкалы от руководства HRS/EHRA/APHRS заключается в возможности постановки диагноза СБ в случаях лекарственно-спровоцированной элевации сегмента ST I типа в сочетании с наличием одного из следующих дополнительных клинических критериев: задокументированная фибрилляция же-лудочков/полиморфная желудочковая тахикардия, аритмогенный обморок, ВСС в семье у лиц моложе 45 лет с негативным заключением аутопсии, Бругада-паттерн сводчатого типа у членов семьи или ночное агональное дыхание. СБ, согласно шанхайской шкале, может быть диагностирован при наличии хотя бы одного ЭКГ-критерия и при общей сумме баллов 3,5 и более. При сумме баллов от 2 до 3 диагноз расценивается как возможный, а сумма баллов менее 2 указывает на отсутствие СБ [2].
Следует различать электрокардиографический феномен СБ и само заболевание. Феноменом СБ следует считать случаи, когда при устранении причинных факторов ЭКГ нормализуется, генетическое тестирование отрицательное, а лекарственная проба не вызывает модификацию ЭКГ по типу Бругада-паттерна. В этом случае Бругада-паттерн I типа может быть спровоцирован, например, ишемией миокарда, метаболическими нарушениями и деформацией грудной клетки (воронкообразная грудная клетка). Эти состояния могут быть расценены как случаи фенокопии. Разница в терминологии необходима, чтобы отличить эти случаи от типичных случаев СБ, поскольку они чаще всего не имеют генетической этиологии. Однако эта номенклатура остается спорной, поскольку темп генотипирования для редких вариантов генов, за исключением SCN5A, на данный момент весьма низок, что ограничивает способность исключать основную генетическую предрасположенность [13].
Регистрация ЭКГ у детей и подростков с наследственными аритмиями не всегда означает своевременную постановку диагноза. Это может быть обусловлено сложностью ее правильной интерпретации и отсутствием характерных для данных синдромов изменений на ЭКГ в состоянии покоя и классическом расположении электродов грудных отведений. Фебрильная температура тела - дополнительный путь поиска пациентов с наследственными нарушениями ритма сердца. Ввиду того, что инфекционные заболевания очень распространены, в том числе среди детей, и инфекции сопровождаются повышением температуры тела, появляется дополнительная возможность выявления больных с наследственными аритмиями. Таким образом, в условиях инфекционного стационара вероятность выявления данного жизнеугрожающего синдрома гораздо больше при условии, что ЭКГ было выполнена на фоне лихорадки (особенно при необъяснимых синкопальных состояниях в анамнезе, случаях ВСС в семье).
Генетическое тестирование на врожденный СБ рекомендовано выполнять всем детям и подросткам при наличии специфических изменений ЭКГ и следующих факторов риска ВСС: эпизоды синкопе, сердечный «арест» в анамнезе, желудочковые нарушения ритма, случаи ВСС в семье [8].
В серии наблюдений Л.А. Бокерия был выявлен ряд особенностей, которые могут указывать на носителей мутаций в гене SCN5A: увеличение интервалов проводимости на ЭКГ покоя (PQ и QRS) и увеличение интервалов АН и HV по данным электрофизиологического исследования. Эти изменения возникают при мутации натриевых каналов по типу полной потери функции, однако выраженность симптомов СБ у данной группы пациентов различна -от бессимптомного носительства до рецидивирующих обмороков, прогрессивных дефектов проводимости и синдрома слабости синусового узла [18].
Следует также отметить электрофизиологические предикторы злокачественных аритмий, такие как дисфункция синусового узла, внутрижелудочковая задержка проводимости, предсердные аритмии, так как аритмогенный субстрат при СБ не должен ограничиваться желудочковым уровнем и вполне может также учитываться к возникновению суправентрикулярных аритмий. Кроме того, наличие глубокого зубца S в первом отведении ЭКГ, по данным Y. Michowitz и соавт., может также указывать на высокую вероятность развития у пациента аритмических событий [12].
Расширяет возможности диагностики применение 24-часового мониторирования по Холтеру, с помощью которого вероятность зарегистрировать ЭКГ-паттерны СБ I типа значительно возрастает в связи с регистрацией ЭКГ в промежутки времени, когда изменения появляются значительно чаще, например, во время сна или после приема пищи. Сравнение чувствительности различных вариантов записи ЭКГ, проведенное K. Shimeno и соавт., продемонстрировало увеличение чувствительности диагностики - от 25% при проведении однократной ЭКГ в 12 отведениях до 38% при 24-часовом мониторировании и до 55% при 24-часовом мониторировании с высоким расположением отведений [19].
Сообщалось также о пользе использования имплантируемого петлевого регистратора для выявления скрытых аритмий, а также для точной оценки симптоматических аритмий. В исследовании D. Righi и соавт. регистратор был безопасно имплантирован без побочных эффектов, что подтверждает пользу этого инструмента в управлении лечением и выборе терапии [5].
Оценка большинства предикторов злокачественных аритмий и стратификация риска бессимптомных пациентов на сегодняшний день вызывают значительные затруднения. Используется шкала для стратификации риска, созданная J. Sieira и соавт. Она включает 6 факторов риска развития злокачественных нарушений сердечного ритма, каждому из которых соответствует определенное количество баллов. Предикторами высокого риска в данной шкале выступают такие симптомы, как обмороки, спонтанный ЭКГ-паттерн I типа, семейный анамнез ВСС, синдром слабости синусового узла, перенесенная ВСС, желудочковые тахиаритмии, индуцируемые электрофизиологическим исследованием. Риск развития аритмии оценивается по сумме набранных баллов: чем она больше, тем выше риск летальных аритмий [20]. Для оценки риска развития жизнеугрожающих аритмий у детей чаще используется специальная педиатрическая шкала, разработанная в 2018 г. Gonzalez Corcia и соавт. Максимальное количество баллов (4) отдается пациентам с симптоматической формой СБ (ВСС в анамнезе, обморок), 3 балла - за спонтанный ЭКГ-паттерн I типа, 2 балла - при дисфункции синусового узла и/или предсердной тахикардии, 1 балл - при нарушениях проводимости. При сумме баллов более 6 риск развития злокачественных аритмических событий превышает 50%, а у больных с суммой баллов 4 и более достигает 30% [2].
Однако ряд исследователей считает, что постановка диагноза СБ у бессимптомных пациентов, имеющих изолированные ЭКГ-признаки Бругада-паттерна I типа, и ожидание у них высокого риска развития ВСС до манифестации характерных симптомов не совсем уместны, так как ежегодная частота злокачественных аритмий у пациентов с изолированным электрокардиографическим СБ составляет 0,5%, а у пациентов с обмороками в анамнезе - 1,9%. Данным пациентам рекомендовано лечение лихорадки, отказ от препаратов, провоцирующих ЭКГ I типа, и наблюдение до манифестации симптомов. С другой стороны, исследование F. Sacher и соавт. показало наличие мотивированных срабатываний имплантируемого кардиовертера-дефибриллятора у 12% пациентов с бессимптомным течением СБ. В многоцентровом исследовании FINGER частота аритмий у бессимптомных пациентов со спонтанным ЭКГ-паттерном I типа в 2 раза превысила частоту аритмий у бессимптомных пациентов с лекарственно-индуцированными изменениями на ЭКГ и составила 0,8 и 0,4% в год соответственно [21]. Таким образом, рассчитанная в соответствии со шкалами стратификации риска ожидаемая вероятность развития летальных аритмий оказалась значительно выше, чем реальная частота их развития в ряде проведенных наблюдений.
Пациенты с обмороками в анамнезе находятся в промежуточной группе риска. Риск летальных аритмических событий в данном случае выше, чем риск у изначально бессимптомных пациентов, но ниже, чем у пациентов с остановкой сердца. Выделяют 2 различные группы по типу обмороков: первая - с аритмическим синкопе и менее благоприятным прогнозом, вторая - с вазовагальным синкопе и хорошим прогнозом. Для грамотной стратификации риска в данном случае важно знать причину потери сознания. В серии французских исследований 57 пациентов с синкопальными состояниями и СБ синкопальное событие было оценено как вероятно аритмическое в 40% случаев, вероятно вазовагальное в 30% и обморок неясного генеза также в 30% случаев. Фибрилляция желудочков во время наблюдения развилась у 22% пациентов из группы с вероятно аритмическим обмороком. Аналогично в голландском исследовании, включавшем 118 пациентов, фибрилляция желудочков развилась лишь у 12% пациентов группы с предположительно аритмическим обмороком [21]. Предположить тип обморока можно по клиническим проявлениям на момент потери сознания. Стоит помнить, что продромы с затуманенным зрением предшествуют неаритмическому обмороку, а аномальное дыхание указывает на аритмический обморок. Недержание мочи или сердцебиение являются менее специфичными симптомами [22]. Однако так как пациенты с СБ демонстрируют более высокий уровень парасимпатической активации и меньший уровень симпатической активации, можно предположить сосуществование различных типов обмороков у пациентов с СБ. Таким образом, вазовагальный тип обморока не исключает СБ [5].
Заключение
Диагностика СБ в детском возрасте затруднена из-за бессимптомного течения, отсутствия изменений на ЭКГ в состоянии покоя и недостаточно точной интерпретации результатов исследований. Необходимо тщательно собирать анамнез, уточняя наличие эпизодов синкопальных состояний, аритмий, особенно при выявлении случаев ВСС у родственников до 45 лет. Таким детям необходимо проводить ЭКГ в высоких грудных отведениях и провокационные пробы для исключения СБ, однако при оценке проб важно принимать во внимание относительно низкую их специфичность. Следует помнить о дополнительной возможности постановки диагноза у пациентов во время лихорадки и при оценке ЭКГ, проведенной во время сна или принятия пищи. Кроме того, возможно использование имплантируемого петлевого регистратора для оценки характера аритмий. Важно своевременно направлять пациентов на генетическое исследование ввиду более высокого риска злокачественных аритмических событий у детей с подтвержденной мутацией SCN5A.
Литература
1. Чупрова С.Н., Руднева Е.П., Лобзин Ю.В. Наследственные нарушения сердечного ритма и проводимости у детей с инфекционными заболеваниями // Медицинский совет. 2020. № 10. С. 126-133.
2. Синдром Бругада. Особенности у детей / В.Н. Федорец, В.С. Василенко, Е.И. Скоробогатова, Н.О. Гончар // Медицина: теория и практика. 2022. № 2. С. 50-61.
3. Worldwide Prevalence of Brugada Syndrome: A Systematic Review and Meta-Analysis / W. Vutthikraivit [et al.] // Acta Cardiologica Sinica. 2018. Vol. 34, No. 3. P. 267-277.
4. Gender differences in patients with Brugada syndrome and arrhythmic events: data from a survey on arrhythmic events in 678 patients / A. Milman [et al.] // Heart Rhythm. 2018. Vol. 15, No. 10. P 1457-1465.
5. Clinical characteristics and risk of arrhythmic events in patients younger than 12 years diagnosed with Brugada syndrome / D. Righi [et al.] // Heart Rhythm. 2021. Vol. 18, No. 10. P 1691-1697.
6. Fibrosis, connexin-43, and conduction abnormalities in the Brugada syndrome / K. Nademanee [et al.] // Journal of the American College of Cardiology. 2015. Vol. 66, No. 18. P 1976-1986.
7. Relationship between arrhythmogenic right ventricular cardiomyopathy and brugada syndrome: new insights from molecular biology and clinical implications / D. Corrado [et al.] // Circulation: Arrhythmia and Electrophysiology. 2016. Vol. 9, No. 4.
8. Синдром Бругада / В.С. Дульченко, А.Х. Магомедова, А.А. Василенко, Л.Д. Хидирова // Евразийский кардиологический журнал. 2020. № 1. С. 130-135.
9. Опыт диагностики и десятилетние результаты лечения пациентов с синдромом Бругада / Л.А. Бокерия [и др.] // Анналы аритмологии. 2017. Т. 14, № 2. С. 60-72.
10. Mutational analysis of mitochondrial DNA in Brugada syndrome / L. Stocchi [et al.] // Cardiovascular Pathology. 2016. Vol. 25, No. 1. P. 47-54.
11. Голубенко М.В., Михайлов В.С., Заклязьминская Е.В. Исследование модифицирующей роли полиморфизма митохондриальной ДНК в манифестации синдрома Бругада // Альманах клинической медицины. 2019. Т. 47, № 1. С. 66-71.
12. Characterization and Management of Arrhythmic Events in Young Patients With Brugada Syndrome / Y. Michowitz [et al.] // Journal of the American College of Cardiology. 2019. Vol. 73. P. 1756-1765.
13. Сергуладзе С.Ю., Проничева И.В. Стратификация риска и критерии диагностики синдрома Бругада: современное состояние и развивающиеся идеи // Анналы аритмологии. 2020. № 1. С. 46-59.
14. Sex Hormone and Gender Difference - Role of Testosterone on Male Predominance in Brugada Syndrome / W. Shimizu [et al.] // Journal of Cardiovascular Electrophysiology. 2007. Vol. 18, No. 4. P. 415-421.
15. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013 / S.G. Priori [et al.] // Heart Rhythm. 2013. Vol. 10, No. 12. P. 1932-1963.
16. The diagnostic yield of Brugada syndrome after sudden death with normal autopsy / M., Papadakis [et al.] // Journal of the American College of Cardiology. 2018. Vol. 71. P. 1204-1214.
17. Wilde A., Antzelevitch C., Borggrefe M. Proposed diagnostic criteria for the Brugada Syndrome // Circulation. 2002. Vol. 106. P. 2514-2519.
18. Бокерия Л.А., Проничева И.В., Сергуладзе С.Ю. Синдром Бругада и перекрестные синдромы сердечной натриевой каналопатии: различные маски мутаций гена SCN5A // Анналы аритмологии. 2018. № 1. С. 40-54.
19. Usefulness of multichannel Holter ECG recording in the third intercostal space for detecting type 1 Brugada ECG: comparison with repeated 12-lead ECGs / K. Shimeno [et al.] // Journal of Cardiovascular Electrophysiology. 2009. Vol. 20, No. 9. P. 1026-1031.
20. A score model to predict risk of events in patients with Brugada Syndrome / J. Sieira [et al.] // European Heart Journal. 2017. Vol. 38, No. 22. P. 1756-1763.
21. Syncope in Brugada syndrome patients: prevalence, characteristics, and outcome / F Sacher [et al.] // Heart Rhythm. 2012. Vol. 9. P. 1272-1279.
22. A score model to predict risk of events in patients with Brugada Syndrome / J. Sieira [et al.] // European Heart Journal. 2017. Vol. 38, No. 22. P. 1756-1763.



