Врожденная патология грудной аорты в педиатрической практике
СтатьиА.С. Шарыкин1, 2, д-р мед. наук, профессор, М.А. Абрамян3, 4, д-р мед. наук, профессор, И.И. Трунина1, 2, д-р мед. наук, профессор, А.Н. Гришкин2
1 ФГАОУ ВО «Российский национальный исследовательский медицинский университет им. Н.И. Пирогова» Минздрава России, г. Москва
2 ГБУЗ «Детская городская клиническая больница им. З.А. Башляевой Департамента здравоохранения г. Москвы»
3 ФГАОУВО «Российский университет дружбы народов им. Патриса Лумумбы», г. Москва
4 ГБУЗ ДЗМ «Морозовская детская городская клиническая больница Департамента здравоохранения г. Москвы»
Резюме. Заболевания грудной аорты проявляются прежде всего ее дилатацией на различных уровнях и несут в себе угрозу развития опасных для жизни осложнений (разрыв, расслоение). Определенные алгоритмы наблюдения за взрослыми больными с дилатацией аорты сформированы и изложены в соответствующих консенсусных рекомендациях. Однако в педиатрической практике определение диаметров аорты, которые можно считать пороговыми для аневризматических изменений, тактика наблюдения за пациентами, лечебные алгоритмы и показания к операции остаются недостаточно изученными. В связи с этим нами выполнен обзор публикаций, посвященных дилатации аорты у детей и подростков.
Ключевые слова: грудная аорта, размер аорты, расслоение аорты, синдромы
Для цитирования: Шарыкин А.С., Абрамян М.А., Трунина И.И., Гришкин А.Н. Врожденная патология грудной аорты в педиатрической практике // Практика педиатра. 2024. № 1. С. 26-34.
Summary. Diseases of the thoracic aorta manifest primarily in its dilation at various levels and carry the threat of developing life-threatening complications (rupture, dissection). Certain algorithms for monitoring adult patients with aortic dilation have been formed and set out in the relevant consensus recommendations. However, in pediatric practice, the determination of aortic diameters, which can be considered thresholds for aneurysmal changes, patient monitoring tactics, therapeutic algorithms and indications for surgery remain insufficiently studied. In this regard, we have undertaken a review of publications on aortic dilation in children and adolescents.
Keywords: thoracic aorta, aortic size, aortic dissection, syndromes
For citation: Sharykin A.S., Abramyan M.A., Trunina I.I., Grishkin A.N. Congenital pathology of the thoracic aorta in pediatric practice // Pediatrician's Practice. 2024;(1):26-34. (In Russ.)
Введение
Врожденные заболевания аорты - это группа патологий, характеризующихся ее дилатацией вплоть до аневризмы c угрозой разрыва или расслоения. Аневризма представляет собой выпячивание кровеносного сосуда, которое обычно возникает там, где стенка стала «слабой», потеряла эластичные свойства и способность возвращаться к своей нормальной форме после растяжения систолическим потоком крови. Однако первоначально можно определить лишь то, что диаметр сосуда превышает значение, ожидаемого для данного возраста и антропометрических данных пациента. Европейские и американские медицинские ассоциации рекомендуют у взрослых лиц констатировать расширение аорты при диаметре >40 мм, а аневризму - при диаметре, превышающем 45-50 мм [1, 2]. Однако в педиатрической популяции для оценки диаметра аорты обычно используется z-score (z-фактор), индексирующий диаметр сосуда по площади поверхности тела, поскольку нормальный размер аорты быстро меняется с возрастом и размерами тела ребенка [3, 4]. Z-score >2,0 считают расширением, а z-score >3,0 - аневризмой аорты для данного возраста и площади поверхности тела. К сожалению, научные работы, посвященные течению острых аортальных синдромов, основаны преимущественно на анализе данных взрослой популяции, редко используют z-score и должны быть переоценены для применения в педиатрической практике. ,
Внимание к аневризме аорты обусловлено угрозой развития опасных для жизни осложнений (разрыв, расслоение). Опасность недиагностированной дилатации заключается в том, что обычно она протекает бессимптомно, а возникшее расслоение может привести к внезапной смерти половины больных [5]. Благодаря накоплению знаний о данной патологии, внедрению ультразвуковых методов исследования и появлению возможности визуализации всех отделов аорты ее ранняя диагностика значительно улучшилась. В связи с этим в настоящее время гигантские аневризмы практически не встречаются, а разрывы вследствие сильного истончения стенки сменились случаями расслоения аорты. Тем не менее точная частота аневризм аорты в популяции неизвестна, так как регистрируются только случаи ее осложнений, в первую очередь диссекции, которая выявляется примерно в 10,4 случая на 100 тыс. пациенто-лет [6]. По данным международного регистра IRAD, острое расслоение в 60-70% случаев возникает в области восходящей аорты [7]. Смертность без операции достигает 58%, после хирургического вмешательства - около 26%.
К настоящему времени определенные алгоритмы наблюдения за взрослыми пациентами с дилатацией аорты сформированы и изложены в соответствующих рекомендациях [1, 2]. Однако вопросы выявления и оценки заболеваний аорты у детей изучены недостаточно полно. В связи с этим нами выполнен обзор публикаций, посвященных дилатации аорты у детей и подростков.
Современные взгляды на причины дилатации грудной аорты
Изменения грудной аорты можно разделить на приобретенные (травматические, дегенеративные) и врожденные. К последним относят как синдромальные (генетические) заболевания, так и формы без четкой идентификации генов.
За последние несколько десятилетий было описано не менее 53 генов, связанных с заболеваниями аорты. Однако в последующем было показано, что непосредственную связь с патологией аорты имеют только 11 из них [8]; наиболее важные мутации затрагивают гены, кодирующие компоненты экстрацеллюлярного матрикса (FBN1, COL3A1,TGFBR1 и TGFBR2), трансформирующего фактора роста (TGFBR1, TGFB2) или контрактильность гладких мышц сосудов (ACTA2, MYH11) [9, 10]. Генетические исследования оказались особенно важными для родственников пробанда, так как позволяют им пройти обследование до того, как возникнут серьезные осложнения.
Заболевания могут обозначаться как несиндромные (если затрагивают только аорту) или синдромные (при сочетании с поражением других органов). Наиболее часто в популяции изменения аорты связаны с синдромом Марфана; следующим по распространенности среди врожденных заболеваний следует двустворчатый аортальный клапан (ДАК). Существуют и другие синдромы, имеющие в своем фенотипе дилатацию аорты: васкулярный вариант синдрома Элерса - Данло, Лойса - Дитца, Тернера, Нунан, Клиппеля - Фейля, cutis laxa, Тричера Коллинза. В табл. 1 приведены наиболее частые заболевания, сочетающиеся с дилатацией аорты.
Таблица 1.
Врожденные синдромы и состояния, сопровождающиеся аневризмой грудной аорты (адаптировано из [2])
| Состояние | Изученные гены | Клинические проявления |
| Синдромальные заболевания грудной аорты* | ||
| Синдром Марфана | FBN1 | Аневризма грудной аорты, ПМК, эктопия хрусталика, характерные изменения скелета |
| Синдром Лойса - Дитца | THFBR1, THFBR2, SMAD3, THFB2, THFB3 | Аневризма грудной аорты и ветвей, диссекция аорты, ПМК, остеоартрит и периферическая нейропатия |
| Синдром Элерса - Данло | COL3A1, FLNA | Аневризма грудной аорты, ПМК, диссекция аорты, пневмоторакс |
| Синдром извитости артерий | SLC2A10 | Извитость больших и средних артерий, аневризма грудной аорты |
| LOX-связанная аневризма грудной аорты | LOX | Аневризма грудной аорты, ДАК, диссекция аорты, марфаноидный вид |
| Синдром дисфункции гладких мышц | ACTA2 | Аневризма грудной аорты, заболевание мозговых сосудов, легочная гипертензия |
| Несиндромные (семейные) заболевания | ||
| Семейный синдром аневризмы и диссекции грудной аорты | ACTA2, MYH11, MYLK, PRKG1, MAT2A, MFAP5, FOXE3, THSD4 | Аневризма грудной аорты, диссекция аорты |
| Аневризма грудной аорты, сочетающаяся с двустворчатым аортальным клапаном | ||
| Семейный двустворчатый аортальный клапан/ аортальный стеноз и аневризма грудной аорты | NOTCH1 | Аневризма грудной аорты, аортальный стеноз |
| Двустворчатый аортальный клапан с аневризмой грудной аорты | THFBR2, MAT2A, GATA5, SMAD6, LOX, ROBO4, TBX20 | Синдромный, несиндромный или семейный вариант |
| Синдром Тернера | XO, Xp | ДАК, коарктация аорты, аневризма грудной аорты, диссекция аорты, лимфедема, перепончатая шея |
* Синдромность зависит от варианта патологического гена у конкретного индивидуума.
Примечание: ДАК - двустворчатый аортальный клапан, ПМК - пролапс митрального клапана.
Из внесиндромных наиболее известны изменения аорты при ДАК. Причем у 20-25% этих пациентов имеются указания на семейные заболевания с измененной растяжимостью и патологически увеличенной аортой у родственников первой степени [11]. Облигатные носители могут иметь ДАК, расширение аорты, оба признака или ни один из них. До последнего времени специфический ген или группа генов в таких семьях точно не идентифицированы. Также не выявлены и другие причины изменений аорты. К примеру, M. Gurwitz и соавт. указывают, что у их пациентов с ДАК корни аорты были значительно шире независимо от наличия значительного стеноза аорты или регургитации. В то же время авторы признают, что у этих пациентов никаких аномальных генетических признаков обнаружено не было [12]. В противовес этому R.T. Hahn и соавт. обнаружили зависимость степени дилатации корня аорты от тяжести гемодинамических нарушений на клапанах, но пришли к выводу, что это только подтверждает гипотезу о том, что один и тот же ген ответственен за аномалии аортального клапана и корня аорты [13].
Факторы риска расслоения аорты
В настоящее время к предикторам формирования аневризмы аорты и ее расслоения относят многочисленные факторы, что отражает генетическую и фенотипическую полиморфность заболевания. Однако не подлежит сомнению роль исходного заболевания и структурной слабости артериальной стенки. Так, при синдроме Марфана основным жизнеугрожающим фактором являются генетически обусловленные и резко выраженные отклонения в строении аортальной стенки, которые приводят к относительно частым операциям уже в детском возрасте [14, 15]. Однако показаниями к хирургическому вмешательству, помимо быстрого расширения корня аорты, считаются и значительная недостаточность аортального клапана, и симптомный пролапс митрального клапана с регургитацией, который является ведущим симптомом синдрома в этом периоде (88%), протекает с выраженными фибромиксоматозными изменениями створок и хорд, растяжением и кальцификацией клапанного кольца и регургитацией более чем в 44% случаев [16, 17].
Риск расслоения аорты повышается при увеличении ее диаметра. В работах [18, 19], специально посвященных этому вопросу, выявлено, что предполагаемый риск диссекции и/или разрыва в течение 5 лет составил 0,4; 1,1 и 2,9% при исходных диаметрах аорты 45, 50 и 55 мм соответственно, т. е. увеличивался по мере усиления дилатации. Риски были низкими при диаметре <5,0 см, если проводилась своевременная плановая пластика аорты, независимо от морфологии аортального клапана. Максимальный диаметр аорты при диссекции составлял 46,7 мм для несиндромных заболеваний и 58,5 мм при синдроме Марфана. Похожие результаты представлены в другой работе [20]: диссекция аорты у пациентов группы риска может возникать, даже если размер аорты нормальный или увеличен минимально (только у 1 из 9 диаметр аорты был ≥5,0 см в корне либо в восходящей аорте).
При ДАК до последнего времени основное внимание уделялось именно клапану, показаниям к его коррекции или протезированию, и считалось, что прогноз во многом зависит от комбинации аневризмы аорты с дисфункцией аортального клапана. Создание соответствующих регистров показало, что дилатация и расслоение аорты возможны и при нормально функционирующем ДАК [21]. Тем не менее было продемонстрировано, что предоперационная тяжелая аортальная регургитация всегда приводит к увеличению частоты повторных операций на аорте [22]. И даже при минимальной аортальной недостаточности через 2-5 лет после реконструкции клапана диаметр аорты способен превысить 99-й центиль для данной площади поверхности тела [14].
Роль гуморальных маркеров дилатации неоднозначна. В одной из работ расширение на уровне восходящей аорты значимо коррелировало с уровнем матриксных металлопротеиназ (ММР2) или белков внеклеточного матрикса (TIMP2) в плазме крови [23]. Хотя из этого исследования остается неясным, являются ли аномальные паттерны кровотока в аорте и экспрессия белка причиной или следствием дилатации аорты, это исследование демонстрирует возможность использования биохимических показателей плазмы в качестве неспецифических предикторов дилатации аорты.
К сожалению, уровень в крови большинства биомаркеров (миозин гладких мышц, кальпонин, растворимые фрагменты эластина, матриксная металлопротеиназа-9, D-димер и др.) увеличивается уже при возникновении диссекции аорты, а не как ее предвестник [24].
Особенности течения наиболее частых заболеваний, сочетающихся с дилатацией аорты
Синдром Марфана. Данный синдром (OMIM 154700) является аутосомно-доминантным заболеванием соединительной ткани с мультисистемными проявлениями, вызванными гетерозиготной мутацией в гене фибриллина-1 (FBN1; OMIM 134797) в локусе 15q21. Заболевание поражает скелетно-мышечную (чрезмерный рост длинных костей, гипермобильность суставов, сколиоз, деформация грудной мышцы), зрительную (близорукость, эктопия хрусталика) и сердечно-сосудистую системы (расширение проксимального отдела восходящей аорты, расширение легочного ствола, пролапс атриовентрикулярных клапанов, аневризмы мозговых артерий). По разным оценкам, распространенность этой патологии составляет от 1 : 3000 до 1 : 5000 [25, 26]. Модифицированные гентские критерии диагностики основываются в основном на наличии дилатации аорты, эктопии хрусталика и мутации фибриллина-1 [27]. Вспомогательную роль играют балльная оценка системных аномалий и семейный анамнез.
Заболевание связано с дефектом гена, который продуцирует фибриллин - белок соединительной ткани. Гистологические особенности медиального слоя корня аорты включают кистозный медиальный некроз, фиброз и утрату гладкомышечных клеток [28, 29]. Аневризма аорты локализуется преимущественно в области ее корня (синусов Вальсальвы), но может затрагивать и восходящую часть. Снижение ожидаемой длительности жизни происходит в первую очередь из-за осложнений со стороны аорты, ее расширения и расслоения. Средняя продолжительность жизни составляет 32,0 ± 16,4 года [30]. Y. Kunita приводит 1 собственный случай и 5 случаев расслоения аорты из литературы, возникших у детей до 14 лет [31]. Таким образом, случаи ранних осложнений вполне вероятны, и вопросы ранней диагностики синдрома, в том числе дилатации аорты, являются весьма актуальными. Следует заметить, что в раннем детском возрасте синдром может манифестировать преимущественно недостаточностью митрального клапана, которая является основной причиной хирургического вмешательства [17].
Превентивное протезирование измененной аорты предупреждает ее диссекцию и улучшает выживаемость пациентов. Показаниями к раннему хирургическому вмешательству могут быть семейная история расслоения, дилатации аорты >2 мм в год, тяжелая недостаточность аортального клапана с расширением левого желудочка.
Лица с двустворчатым аортальным клапаном из-за высокой частоты последнего в популяции составляют основной контингент с расширенной аортой в детском возрасте. Несмотря на то, что некоторые крупные серии вскрытий при расслоении аорты не выявили ее исключительной связи с ДАК [32], он, тем не менее, вошел в перечень факторов риска аномалий стенки аорты наряду с сопутствующей клапанной дисфункцией, коарктацией аорты или артериальной гипертензией. При этом выдвигавшаяся гипотеза о кистозном медианекрозе, ослабляющем стенку аорты, несмотря на ее привлекательность [33], оказалась недостаточно состоятельной. Было показано, что кистозный медианекроз может наблюдаться у пожилых пациентов без серьезных аномалий аорты и отсутствовать у других пациентов с явными признаками хрупкости стенки аорты [33]. С большей уверенностью можно говорить о потере эластичных пластинок и фиброзе интимы [6].
Значительное количество исследований посвящено связи дисфункции двустворчатого клапана с дилатацией аорты из-за нарушений гемодинамики, вызванных эксцентричностью и деформацией потока крови. Однако изолированная дисфункция клапана, по-видимому, является недостаточным объяснением патологии аорты. Так, R.T.Pachulski и соавт. показали, что диаметр синусов Вальсальвы при ДАК без стеноза или регургитации был все равно больше, чем в контрольной группе [34]. Сходные выводы были сделаны в другом исследовании, при котором у 110 (66%) из 167 пациентов с ДАК дисфункция аортального клапана присутствовала, но степень дилатации не коррелировала с тяжестью аортальной недостаточности [35].
С появлением магнитно-резонансной томографии (МРТ) большее внимание стали уделять степени эксцентричности экскурсии створок, изменяющей направление трансклапанного кровотока. В работе P.M. den Reijer и соавт. обнаружены значимые корреляции между углом струи кровотока с осью аорты и диаметрами аорты на уровне синусов Вальсальвы, синотубулярного соединения или восходящей аорты. Большему углу неправильно направленного потока соответствовал больший диаметр аорты. Корреляции были наиболее значимы на более дистальном уровне восходящей аорты, где напряжение стенки и давление ожидаемо самые высокие [23].
Величина расширения восходящей аорты среди лиц с ДАК колеблется от 20 до 84% [36]. Эта вариабельность связана с различиями в исследуемых популяциях, методах оценки и выбранных пороговых значениях размера аорты. Для описания двустворчатой аортопатии были предложены схемы классификации, основанные на гистологических особенностях, морфологических моделях сращивания клапанов и иерархическом кластерном анализе, но ни одна из них не получила широкого распространения [37, 38].
Следует особо отметить, что для детей ссылки на «взрослые» диаметры аорты неприемлемы. Необходимо рассчитывать измеренные показатели с поправкой на площадь поверхности тела и представлять их в виде z-score. Так, P.M. den Reijer и соавт. при МРТ-исследовании детей с ДАК в возрасте 15,0 ± 1,9 года. и средней площади поверхности тела 1,7 ± 0,3 м2 установили, что средний диаметр синусов Вальсальвы равнялся 33,1 ± 1,0 мм, а восходящей аорты - 30,8 ± 1,2 мм, что соответствует z-score 2,27 и 3,0 соответственно. Таким образом аневризматическое расширение аорты было выявлено в 77,8% случаев [23].
Пациентам с ДАК, с расширением аорты или без него, требуется пожизненное наблюдение за корнем аорты и восходящей аортой из-за риска непредсказуемого роста аорты [2]. Так, в когорте взрослых пациентов с ДАК без дилатации аорты на исходном этапе (<4,5 см) через 14 ± 6 лет после постановки диагноза у 13% развилась аневризма аорты, а 25-летний риск составил 26%. Авторы делают вывод, что превышение диаметра в 4 см должно трактоваться как дилатация аорты и тщательно наблюдаться. По данным K.W. Holmes и соавт. необходимость вмешательств на аорте нередко возникает уже в первую декаду жизни. В соответствии с представленной ими актуарной кривой к возрасту 30 лет элективно оперируют уже 3,3% пациентов с ДАК (и диагностированной дилатацией аорты), а к 40 лет - 8,3% [39].
Лечебные алгоритмы
Медикаментозная терапия. Начиная с 90-х гг. XX в. для профилактики интенсивной дилатации аорты при синдроме Марфана стали применять бета-блокаторы [40]. В последующем экспериментальные модели с использованием антител, нейтрализующих трансформирующий фактор роста бета (TGFb), и лозартана, показали впечатляющие результаты, не только снижая рост корня аорты до нормального уровня, но и ингибируя фрагментацию эластических волокон в стенке аорты [41]. С тех пор обе группы препаратов используют с целью профилактики формирования аневризмы аорты. Однако результаты лечения значительно варьируют в разных исследованиях. Так, в одной работе был показан меньший прирост размера аорты в течение 3,5 года у больных, получавших лозартан (151 чел.), по сравнению с контрольной группой (148 чел.), однако недостоверный [42]. Примерно равным было и количество хирургических вмешательств в этих группах (15 и 13 соответственно). Добавление к лечению статинов также показало лишь ограниченную эффективность в борьбе с аневризмами аорты [43]. Однако последние передовые технологии в области анализа отдельных клеток, протеомики и искусственного интеллекта открывают новые возможности для идентификации различных терапевтических мишеней, участвующих в клеточной гетерогенности, воспалении сосудов, оксидативном стрессе, гибели клеток, интрамуральном тромбозе. Потенциально новым направлением может быть воздействие на митохондриальные дефекты в клетках аорты [44].
К сожалению, до последнего времени не продемонстрировано существенного профилактического эффекта известных медикаментов, и все еще неясно, может ли его оказывать какой-либо существующий препарат [45].
Модификация образа жизни. Небольшие аневризмы несут низкий риск, поэтому рекомендуется тщательное наблюдение за их ростом, контроль артериального давления, отказ от курения, ограничения физических нагрузок.
Руководство ESC по спортивной кардиологии (2020) содержит конкретные рекомендации в отношении назначения физической активности пациентам с синдромом Марфана или другими заболеваниями аорты [46]. Наиболее подробно описаны рекомендации при синдроме Марфана, представленные ниже.
1. Пациенты с отсутствием дилатации аорты (т. е. с максимальным диаметром аорты <40 мм) относятся к группе низкого или среднего риска, но им рекомендуется избегать упражнений высокой и очень высокой интенсивности, контактных и силовых дисциплин, а повторное клиническое обследование пациента проводят каждые 1-2 года.
2. Пациентов с умеренной дилатацией аорты (т. е. с максимальным диаметром аорты 40-45 мм) относят к группе среднего риска, и у этих людей разрешены только смешанные или связанные с выносливостью дисциплины низкой интенсивности с периодическим наблюдением каждые 6-12 мес.
3. Пациенты с выраженной дилатацией аорты (т. е. с максимальным диаметром аорты >45 мм) относятся к группе высокого риска, и им противопоказаны все виды спорта.
Вопрос, оправданны ли данные рекомендации для юных пациентов с ДАК, подлежит дальнейшему изучению.
Роль хирургического вмешательства. Более радикальный лечебный подход оценен в исследовании «Эффективное лечение аневризм грудной аорты» (ETTAA), охватившем 886 пациентов [47]. Были изучены темпы роста аневризм, исходы у пациентов, качество жизни и затраты, в том числе связанные с хирургическим вмешательством. Сравнивались 3 основных метода: длительное тщательное наблюдение, хирургическая коррекция на открытом сердце и стентирование.
В общей сложности 489 пациентов подверглись тщательному наблюдению, 112 получили консервативное лечение. Без хирургического вмешательства показатели смертности были выше у женщин и пожилых пациентов, при этом риск смерти удваивался на каждый сантиметр диаметра аневризмы в исходном состоянии. Исходный диаметр аневризмы был выше у пожилых пациентов, курильщиков, пациентов с соединительнотканной дисплазией, хронической обструктивной болезнью легких или пороком клапанов - при факторах, которые необходимо оценивать при планировании лечения. Из оставшихся пациентов 150 перенесли стентирование, а 135 - открытую операцию. Общая годичная выживаемость составила 83% после имплантации стента и 79% после открытой операции, но разница могла быть обусловлена случайностями, а стентированию подвергалась только нисходящая аорта.
Авторы делают вывод, что при больших аневризмах важно, чтобы операция не откладывалась, так как более длительное ожидание операции приводит к ухудшению результатов.
Оперативные вмешательства в молодом возрасте
С появлением доказательств врожденного характера расширения аорты возникла необходимость изучить данную проблему у молодых пациентов и детей. Как оказалось, в педиатрической практике наибольшее число пациентов с проявлениями дилатации аорты имеет синдром Марфана и ДАК [39, 48], которые, помимо угроз для жизни, могут ухудшать качество жизни. Дети с соединительнотканными синдромами часто сообщают о боли и усталости, ухудшении общего и ментального здоровья, ограничениях активности и участия в социальной жизни [49].
Что касается необходимости хирургического вмешательства при аневризме аорты, то наибольшим опытом наблюдения за такими пациентами располагает генетический регистр в США, охватывающий 8 клинических центров [39]. По их данным, за 8 лет элективные и экстренные хирургические вмешательства были проведены 896 (33,3%) из 2686 лиц в 6 основных диагностических группах (табл. 2).
Таблица 2.
Исходы врожденных заболеваний грудной аорты (адаптировано из [39])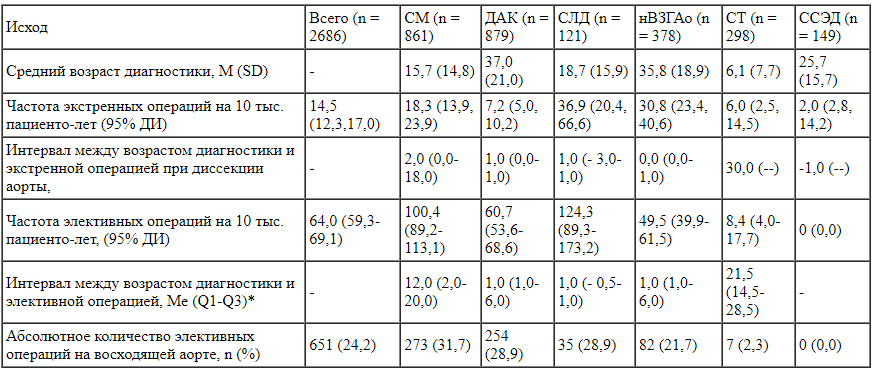 * Положительные различия указывают на то, что диагноз был поставлен до события, а отрицательные - что диагноз был поставлен после события.
* Положительные различия указывают на то, что диагноз был поставлен до события, а отрицательные - что диагноз был поставлен после события.
Примечание: СМ - синдром Марфана, ДАК - двустворчатый аортальный клапан, СЛД - синдром Лойса - Дитца, нВЗГАо - несиндромные врожденные заболевания грудной аорты, СТ - синдром Тернера, ССЭД - сосудистый синдром Элерса - Данло.
Исходя из представленных данных, можно сделать вывод о том, что синдромы с хорошо выраженными фенотипическими признаками (синдромы Марфана, Лойса - Дитца, Тернера) диагностируются в более молодом возрасте, чем остальные заболевания. Последние, вероятно, были зарегистрированы только при появлении клинической картины или при обследовании с применением методов визуализации. Это подтверждается и коротким интервалом между диагностикой и последующей экстренной или плановой операцией (в среднем 1 год в противовес 2-12 годам при синдроме Марфана).
Наиболее часто неотложные операции выполняют при синдроме Лойса - Дитца и несиндромных врожденных заболеваниях грудной аорты, однако частота этих заболеваний в популяции относительно невелика. Большего внимания требуют пациенты с синдромом Марфана и ДАК. Конечно, основные риски представляет синдром Марфана, при котором происходит подавляющая часть диссекций аорты (61% по данным [20]). Совокупная частота диссекции среди этих пациентов в 6 раз выше (4,5%), чем среди остальных лиц (0,7%; р < 0,001), в том числе среди имеющих ДАК (0,3%; p < 0,001). Однако представленность ДАК в популяции составляет 2% (в 100 раз больше, чем синдрома Марфана), что делает его сопоставимым с указанным синдромом по абсолютному количеству случаев дилатации аорты.
Что касается плановых операций по удалению аневризмы аорты, они проводятся при синдромах Марфана и Лойса - Дитца с относительно высокой частотой в раннем и среднем возрасте (по сравнению с операциями по устранению расслоения аорты) [39], а для ДАК характерна высокая частота плановых операций в любом возрасте.
Своевременные элективные вмешательства для предотвращения диссекции аорты, проводимые специализированными хирургическими бригадами, являются, по-видимому, наиболее важным фактором долгосрочной выживаемости, учитывая значительное снижение послеоперационной летальности в последние годы (2-4% среди пациентов с синдромом Марфана и ДАК) [39].
Заключение
Расслоение аорты - опасное для жизни, но потенциально предотвратимое состояние. Аневризма аорты у взрослых не возникает внезапно. В связи с этим предпринимаются попытки установить закономерности прогрессирования заболевания при наличии предрасположенности к нему, как при синдроме Марфана, так и у детей с ДАК. Ранняя диагностика, лечение и тщательное наблюдение имеют решающее значение для выживания. Учитывая врожденный характер патологии, ее выявление возможно уже в детском возрасте, однако алгоритмы диагностики и последующего наблюдения фактически отсутствуют.
Существуют различные методы визуализации расширенной аорты - рентгенологический, эхокардиографический, с помощью компьютерной томографии или МРТ. Последние являются наиболее точными, допускающими объемную реконструкцию изображения на всем протяжении аорты. Однако они относительно дороги и малопригодны, так как требуют длительного неподвижного состояния детей, в связи с чем приходится прибегать к их глубокой седации. Наиболее востребованной в детской практике является эхокардиографическая диагностика. Современные ультразвуковые приборы обладают хорошими разрешающими способностями, позволяя исследовать грудную аорту на всем ее протяжении, в том числе повторно. Однако для оценки роста аорты при динамических наблюдениях на протяжении длительного времени необходимо использовать нормативные педиатрические базы по диаметрам аорты, ориентированные не на возраст, а на площадь поверхности тела.
Возможности профилактики формирования аневризмы аорты в настоящее время ограничены. Применение бета-блокаторов или блокаторов рецепторов АПФ находится на стадии накопления больших объемов наблюдений за длительные периоды. Учитывая сообщения о влиянии на состояние аорты повышенного артериального давления, физических нагрузок, курения, относительно перспективным представляется формирование правильного образа жизни.
Поскольку реализация генетически предопределенного дефекта аорты происходит в значительной степени индивидуально, необходимо формировать алгоритмы оценки статуса аорты в зависимости от тех факторов риска, которые имеются у конкретного подростка.
Литература
1. 2014 ESC Guidelines on the diagnosis and treatment of aortic diseases: Document covering acute and chronic aortic diseases of the thoracic and abdominal aorta of the adult. The Task Force for the Diagnosis and Treatment of Aortic Diseases of the European Society of Cardiology (ESC) / R. Erbel [et al.] // European Heart Journal. 2014. Vol. 35, No. 41. P. 2873-2926.
2. 2022 ACC/AHA Guideline for the Diagnosis and Management of Aortic Disease: A Report of the American Heart Asso-ciation/American College of Cardiology Joint Committee on Clinical Practice Guidelines / E.M. Isselbacher [et al.] // Circulation. 2022. Vol. 146, No. 24. P. e334-e482.
3. Sluysmans T., Colan S.D. Theoretical and empirical derivation of cardiovascular allometric relationships in children // Journal of Applied Physiology. 1985. Vol. 99, No. 2. P. 445-457.
4. Nomograms for two-dimensional echocardiography derived valvular and arterial dimensions in Caucasian children / M.Cantinotti [et al.] // Journal of Cardiology. 2017. Vol. 69, No. 1. P. 208-215.
5. Population-based study of incidence and outcome of acute aortic dissection and premorbid risk factor control: 10-year results from the Oxford Vascular Study / D.P. Howard [et al.] // Circulation. 2013. Vol. 127, No. 20. P. 2031-2037.
6. Saratzis A., Bown M.J. The genetic basis for aortic aneurysmal disease // Heart. 2014. Vol. 100. P. 916-922.
7. The International Registry of Acute Aortic Dissection (IRAD): new insights into an old disease / P.G. Hagan [et al.] // JAMA. 2000. Vol. 283, No. 7. P. 897-903.
8. Fatigue in patients with syndromic heritable thoracic aortic disease: a systematic review of the literature and a qualitative study of patients' experiences and perceptions / G. Velvin, H. Johansen, A. 0stertun-Geirdal, T. Bathen // Orphanet Journal of Rare Diseases. 2023. Vol. 18, No. 1. P. 119.
9. Pyeritz R.E. Heritable thoracic aortic disorders // Current Opinion in Cardiology. 2014. Vol. 29, No. 1. P. 97-102.
10. Association Between Genetic Diagnosis and Clinical Outcomes in Patients With Heritable Thoracic Aortic Disease / T. Yagyu [et al.] // Journal of the American Heart Association. 2023. Vol. 12, No. 8. P e028625.
11. Silberbach M. Bicuspid aortic valve and thoracic aortic aneurysm: toward a unified theory // Journal of the American College of Cardiology. 2009. Vol. 53, No. 24. P. 2296-2297.
12. Frequency of aortic root dilatation with a bicuspid aortic valve / M. Gurwitz, R.K. Chang, S. Drant, V Allada // The American Journal of Cardiology. 2004. Vol. 94. P. 1337-1340.
13. Association of aortic dilatation with regurgitant, stenotic and functionally normal bicuspid aortic valves / R.T. Hahn, M.J. Roman, A.H. Mogtador, R.B. Devereux // Journal of the American College of Cardiology. 1992. Vol. 19. P. 283-288.
14. Surgery for aortic root aneurysm in children: a 21-year experience in 50 patients / S.M. Cattaneo [et al.] // Annals of Thoracic Surgery. 2004. Vol. 77, No. 1. P 168-176.
15. Медикаментозная терапия при синдроме Марфана / A. С. Шарыкин [и др.] // Педиатрия. 2018. Т. 97, № 1. С. 114-121.
16. Natural history of cardiovascular manifestations in Marfan syndrome / C.D. van Karnebeek [et al.] // Archives of Disease in Childhood. 2001. Vol. 84. P 129-137.
17. Неонатальная форма синдрома Марфана - клиническое описание и комплексный подход к диагностике и лечению / B. А. Румянцева, Ю.А. Рогожина, Н.П. Котлукова, E.B. Заклязьминская // Российский кардиологический журнал. 2014. № 5 (109). С. 55-60.
18. Aortic diameter predicts acute type A aortic dissection in patients with Marfan syndrome but not in patients without Marfan syndrome / E.K. Kim [et al.] // Journal of Thoracic and Cardiovascular Surgery. 2014. Vol. 147, No. 5. P. 1505-1510.
19. Risk of Aortic Dissection in the Moderately Dilated Ascending Aorta / J.B. Kim [et al.] // Journal of the American College of Cardiology. 2016. Vol. 68, No. 11. P 1209-1219.
20. Aortic Dissection in Patients With Genetically Mediated Aneurysms: Incidence and Predictors in the GenTAC Registry / J.W. Weinsaft [et al.] // Journal of the American College of Cardiology. 2016. Vol. 67, No. 23. P 2744-2754.
21. Characterizing the young patient with aortic dissection: results from the International Registry of Aortic Dissection (IRAD) / J.L. Januzzi [et al.] // Journal of the American College of Cardiology. 2004. Vol. 43, No. 4. P 665-669.
22. Reported Outcome After Valve-Sparing Aortic Root Replacement for Aortic Root Aneurysm: A Systematic Review and Meta-Analysis / B. Arabkhani [et al.] // Annals of Thoracic Surgery. 2015. Vol. 100, No. 3. P 1126-1131.
23. Hemodynamic predictors of aortic dilatation in bicuspid aortic valve by velocity-encoded cardiovascular magnetic resonance / P.M. den Reijer [et al.] // Journal of Cardiovascular Magnetic Resonance. 2010. Vol. 12, No. 1. P 4.
24. The IRAD and beyond: what have we unravelled so far? / X. Yuan, A. Mitsis, Y. Tang, C.A. Nienaber // General Thoracic and Cardiovascular Surgery. 2019. Vol. 67, No. 1. P. 146-153.
25. Judge D.P., Dietz H.C. Marfan's syndrome // Lancet. 2005. Vol. 366, No. 9501. P 1965-1976.
26. Inherited Thoracic Aortic Disease: New Insights and Translational Targets / A.J. Fletcher [et al.] // Circulation. 2020. Vol. 141, No. 19. P 1570-1587.
27. The revised Ghent nosology forthe Marfan syndrome / B.L. Loeys [et al.] // Journal of Medical Genetics. 2010. Vol. 47. P 476-485.
28. Schlatmann T.J., Becker A.E. Histologic changes in the normal aging aorta: implications for dissecting aortic aneurysm // The American Journal of Cardiology. 1977. Vol. 39, No. 1. P. 13-20.
29. Schlatmann T.J., Becker A.E. Pathogenesis of dissecting aneurysm of aorta. Comparative histopathologic study of significance of medial changes // The American Journal of Cardiology. 1977. Vol. 39, No. 1. P 21-26.
30. Life expectancy and causes of death in the Marfan syndrome / J.L. Murdoch [et al.] // The New England Journal of Medicine. 1972. Vol. 286, No. 15. P 804-808.
31. Kunita Y. A pathological study of dissecting aneurysm in individuals under forty years of age including a case of a 13 years old boy // Acta Pathologica Japonica. 1978. Vol. 28, No. 2. P 253-263.
32. Wilson S.K., Hutchins G.M. Aortic dissecting aneurysms: causative factors in 204 subjects // Archives of Pathology & Laboratory Medicine. 1982. Vol. 106, No. 4. P. 175-180.
33. Lindsay J. Jr. Coarctation of the aorta, bicuspid aortic valve and abnormal ascending aortic wall // The American Journal of Cardiology. 1988. Vol. 61, No. 1. P. 182-184.
34. Pachulski R.T., Weinberg A.L., Chan K.L. Aortic aneurysm in patients with functionally normal or minimally stenotic bicuspid aortic valve // The American Journal of Cardiology 1991. Vol. 67, No. 8. P 781-782.
35. Frequency of bicuspid aortic valve in young male conscripts by echocardiogram / S. Nistri [et al.] // The American Journal of Cardiology. 2005. Vol. 96. P 718-721.
36. Verma S., Siu S.C. Aortic dilatation in patients with bicuspid aortic valve // The New England Journal of Medicine. 2014. Vol. 370, No. 20. P 1920-1929.
37. The aortopathy of bicuspid aortic valve disease has distinctive patterns and usually involves the transverse aortic arch / S.S. Fazel [et al.] // Journal of Thoracic and Cardiovascular Surgery. 2008. Vol. 135, No. 4. P 901-907, 907.e1-2.
38. The American Association for Thoracic Surgery consensus guidelines on bicuspid aortic valve-related aortopathy: Full online-only version / M.A. Borger [et al.] // Journal of Thoracic and Cardiovascular Surgery. 2018. Vol. 156, No. 2. P. e41-e74.
39. Cardiovascular Outcomes in Aortopathy: GenTAC Registry of Genetically Triggered Aortic Aneurysms and Related Conditions / K.W. Holmes [et al.] // Journal of the American College of Cardiology. 2022. Vol. 79, No. 21. P. 2069-2081.
40. Progression of aortic dilatation and the benefit of long-term beta-adrenergic blockade in Marfan's syndrome / J. Shores, K.R. Berger, E.A. Murphy, R.E. Pyeritz // The New England Journal of Medicine. 1994. Vol. 330, No. 19. P. 1335-1341.
41. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome / J.P. Habashi [et al.] // Science. 2006. Vol. 312. P 117-121.
42. Marfan Sartan: a randomized, double-blind, placebo-controlled trial / O. Milleron [et al.] // European Heart Journal. 2015. Vol. 36, No. 32. P 2160-2166.
43. Cho M.J., Lee M.R., Park J.G. Aortic aneurysms: current pathogenesis and therapeutic targets // Experimental & Molecular Medicine. 2023. Dec. 1.
44. Extracellular Tuning of Mitochondrial Respiration Leads to Aortic Aneurysm / J. Oller [et al.] // Circulation. 2021. Vol. 143, No. 21. P 2091-2109.
45. Causes of Mortality in the Marfan Syndrome (from a Nationwide Register Study) / K.A. Groth [et al.] // The American Journal of Cardiology. 2018. Vol. 122, No. 7. P. 1231-1235.
46. 2020 ESC Guidelines on sports cardiology and exercise in patients with cardiovascular disease / A. Pelliccia [et al.] // European Heart Journal. 2021. Vol. 42, No. 1. P. 17-96.
47. Endovascular stent grafting and open surgical replacement for chronic thoracic aortic aneurysms: a systematic review and prospective cohort study / L. Sharples [et al.] // Health Technology Assessment. 2022. Vol. 26, No. 6. P. 1-166.
48. Aortic dilation, genetic testing, and associated diagnoses / Y.A. Zarate [et al.] // Genetics in Medicine. 2016. Vol. 18, No. 4. P 356-363.
49. Heritable connective tissue disorders in childhood: Decreased health-related quality of life and mental health / J. Warn-ink-Kavelaars [et al.] // American Journal of Medical Genetics Part A. 2022. Vol. 188, No. 7. P 2096-2109.



