Фенилкетонурия и нарушения обмена тетрагидробиоптерина у детей. Клинические рекомендации.
Статьи
Фенилкетонурия и нарушения обмена тетрагидробиоптерина у детей
- Союз педиатров России
Оглавление
- Ключевые слова
- Список сокращений
- Термины и определения
- 1. Краткая информация
- 2. Диагностика
- 3. Лечение
- 4. Реабилитация
- 5. Профилактика и диспансерное наблюдение
- 6. Дополнительная информация, влияющая на течение и исход заболевания
- Критерии оценки качества медицинской помощи
- Список литературы
- Приложение А1. Состав рабочей группы
- Приложение А2. Методология разработки клинических рекомендаций
- Приложение А3. Связанные документы
- Приложение Б. Алгоритмы ведения пациента
- Приложение В. Информация для пациентов
- Приложение Г.
Ключевые слова
- Гиперфенилаланинемия
- Гипофенилаланиновая диета
- Классическая фенилкетонурия
- Сапроптерина дигидрохлорид
- Тетрагидробиоптерин
- Тирозин
- Фенилаланин
- Фенилаланин-4-гидроксилаза
Список сокращений
AdGTPCH - Аутосомно-доминантный дефицит гуанозин-трифосфат-циклогидролазы I
ArGTPCH - Аутосомно-рецессивный дефицит гуанозин-трифосфат-циклогидролазы I
ВН4 - кофактор тетрабиоптерин
DHPR - фермент дигидроптеридинредуктаза
GTPСH - фермент гуанозинтрифосфатциклогидролаза
OMIM - on-line mendelian inheritance of man - электронная база данных "Менделевское наследование у человека"
PAH - фермент фенилаланингидроксилаза
PCBD - фермент птерин-4-альфа-карбиноламиндегидратаза
PTPS - фермент 6-пирувоилтетрагидроптеринсинтаза
SR - фермент сепиаптеринредуктаза
ГФА - гиперфенилаланинемия
МГК - медико-генетическая консультация
МРТ - магнитно-резонансная томография
УЗИ - ультразвуковое исследование
ФА - фенилаланин
ФАГ - фермент фенилаланингидроксилаза
ФКУ - фенилкетонурия
ЭЭГ - электроэнцефалография
Термины и определения
«Материнская фенилкетонурия» - эмбриофетопатия, развивающаяся у плода в результате воздействия продуктов аномального метаболизма беременной женщины с фенилкетонурией при отсутствии диетического лечения.
Неонатальный скрининг – медицинская диагностическая технология сплошного безвыборочного лабораторного обследования всех новорожденных на некоторые заболевания обмена веществ, призванная обеспечить своевременное выявление и начало лечения больных детей с целью предотвращения их инвалидизации.
Пренатальная диагностика фенилкетонурии - комплексная дородовая диагностика с целью выявления фенилкетонурии на стадии внутриутробного развития путем определения активности фенилаланингидроксилазы в культуре амниоцитов, биоптате и культуре хориона.
1. Краткая информация
1.1 Определение
Гиперфенилаланинемия (ГФА) – группа аутосомно-рецессивных заболеваний, обусловленных нарушением обмена незаменимой аминокислоты фенилаланина (ФА), поступающей в организм человека с белковой пищей. ГФА объединяет несколько генетически гетерогенных форм нарушения обмена фенилаланина, сходных по клиническим признакам: классическая фенилкетонурия (ФКУ), обусловленная дефицитом фенилаланин-4-гидроксилазы (ФАГ) и гиперфенилаланинемии (ГФА), связанные с нарушением обмена тетрагидробиоптерина (BH4).
1.2 Этиология и патогенез
Фенилкетонурия (в современной классификации - ФАГ зависимая ФКУ) обусловлена дефицитом фермента фенилаланингидроксилазы (ФАГ), приводящим к накоплению в биологических жидкостях фенилаланина (гиперфенилаланинемии) и продуктов его распада. Заболевание вызвано мутацией гена фенилаланингидроксилазы (РАН), локализующегося на длинном плече хромосомы 12, участке 12q22-q24.1.
Нарушения обмена тетрагидробиоптерина – гетерогенная группа гиперфенилаланинемических состояний, вызванная дефицитом одного из ферментов, участвующих в цепи биохимических превращений тетрагидробиоптерина. Дефицит или недостаточная активность ферментов являются результатом мутаций в соответствующих генах.
В норме в организме человека основное количество фенилаланина утилизируется путем превращения его в тирозин, который в свою очередь служит субстратом для синтеза биогенных аминов и меланина. Лишь небольшое количество фенилаланина используется для синтеза белка. Превращение L-фенилаланина в L-тирозин осуществляется с помощью фермента фенилаланингидроксилазы (ФАГ).
В основе патогенеза гиперфенилаланинемий лежит блокирование гидроксилирования фенилаланина и превращения его в тирозин. Прямым следствием нарушения гидроксилирования являются накопление фенилаланина в крови и моче и снижение образования тирозина. У нелеченых лиц с фенилкетонурией и ее вариантами, обусловленными недостаточностью тетрагидробиоптерина, концентрация фенилаланина в плазме достигает уровня, достаточно высокого для активации альтернативных путей метаболизма с образованием фенилпирувата, фенилацетата, фениллактата и других производных, оказывающих токсический эффект на различные органы и ткани (Приложение Г1, рис.1). В наибольшей степени страдают структуры центральной нервной системы.
Выраженное повреждение мозга может быть связано с рядом эффектов избытка фенилаланина: лишением мозга других аминокислот, необходимых для синтеза белка, что объясняется, вероятно, торможением их всасывания в желудочно-кишечном тракте или нарушением реабсорбции из почечных канальцев в условиях избыточного содержания фенилаланина в жидких средах организма, нарушением образования или стабилизации полирибосом, снижением синтеза миелина и недостаточным синтезом норадреналина и серотонина, имеющих исключительно важную роль в созревании и функционировании центральной нервной системы.
Фенилаланин представляет собой конкурентный ингибитор тирозиназы — ключевого фермента на пути синтеза меланина. Блокада этого пути наряду с уменьшением доступности предшественника меланина (тирозина) обусловливает недостаточную пигментацию волос и кожи.
Активность ФАГ зависит от трех основных кофакторов: ФАГС, тетрагидробиоптерина (ВН4), молекулярного кислорода. Наибольшее значение из них имеет тетрагидробиоптерин. Функция этого кофактора заключается в доставке кислорода к активному центру ФАГ, в котором происходит реакция гидроксилирования фенилаланина. Иными словами, тетрагидробиоптерин служит восстановителем для молекулярного кислорода в процессах встраивания его в ряд субстратов (фенилаланин, тирозин, триптофан). Биоптерин-зависимыми монооксигеназами также являются тирозиновая и триптофановая гидроксилазы. Реакции, в которых биоптерин играет роль кофактора, представлены на рисунке 2 (Приложение Г1). В процессе этих реакций кофактор переходит в дигидроформу – дигидробиоптерин.
1.3 Эпидемиология
Частота ФКУ значительно варьирует в зависимости от популяции и составляет от 1:4370 в Турции до 1:80500 в Японии. Наибольшую распространенность заболевание получило у лиц европеоидной расы, однако, и у них его частота существенно варьирует в различных регионах и этнических группах. По данным европейских центров неонатального скрининга, частота заболевания в восточно-европейской популяции выше, чем в популяциях запада и юго-запада Европы. Так, частота ФКУ в Ирландии составляет 1:4500 новорожденных, в Югославии 1:7 300, тогда как в Италии 1:12 280, Греции 1:18 640. В Скандинавских популяциях частота ФКУ исключительно низка, особенно в Финляндии (1:71 000) и Швеции (1:43 230). В России по данным неонатального скрининга частота фенилкетонурии составляет 1:7 000 и колеблется по регионам от 1:4 735 в Курской области до 1:18 000 в Республике Тыва. В Санкт-Петербурге частота ФКУ 1:7 600 новорожденных, в Москве 1:6 772. Наиболее часто встречается классическая форма ФКУ, на долю птерин-зависимых форм приходится 1-3% случаев гиперфенилаланинемий [4,5,10,11,19].
Фенилкетонурия, обусловленная дефицитом фермента РАН, встречается в большинстве случаев гиперфенилаланинемий, выявленных в ходе неонатального скриннга (97-98%).
Гиперфенилаланинемии, связанные с нарушением обмена тетрагидробиоптерина (ранее называемые «атипичная ФКУ») составляют около 2-3% всех случаев ГФА. Эти состояния обусловлены дефицитом различных ферментов, участвующих в синтезе или восстановлении тетрагидробиоптерина (ВН4).
1.4 Кодирование по МКБ-10
Е70.0 Классическая фенилкетонурия
Е70.1 Другие виды гиперфенилаланинемии
1.5 Классификация
Классификация классической фенилкетонурии основана на степени повышения концентрации фенилаланина в крови, определенной до лечения (на скрининге) (табл. 1). До появления данных молекулярно-генетических исследований ГФА считалось, что тяжесть заболевания и степень поражения интеллекта зависят только от уровня ФА в крови, что тесно связано со степенью активности фермента.
Таблица 1. Рабочая классификация ФКУ, обусловленной дефицитом ФАГ, в зависимости от уровня фенилаланина крови до лечения [14]
|
Форма заболевания |
Уровень фенилаланина в сыворотке крови* |
|
|
мкмоль/л |
мг/дл |
|
|
Легкая гиперфенилаланинемия (ГФА, не ФКУ) |
120-600 |
2-10 |
|
Умеренная (мягкая, средняя) ФКУ |
600-1200 |
10-20 |
|
Классическая (тяжелая) ФКУ |
?1200 |
?20 |
* - коэффициент пересчета мкмоль/л в мг/дл равен 60.
На основе результатов молекулярно-генетических исследований создана современная классификация, представленная в медицинской базе данных OMIM, которая хорошо отражает этиопатогенез ГФА и ФКУ (таблица 2).
Таблица 2. Этиопатогенетическая классификация ФКУ и ГФА [14]
|
Код OMIM |
Название патологии |
Фермент |
Ген |
Локализация гена |
|
261600 |
ФАГ зависимая фенилкетонурия (ФКУ, ФАГ дефицит) |
фенилаланин-4-гидроксилаза (РАН) |
РАН |
12q22-q24.2 |
|
Гиперфенилаланинемия, BH4-дефицит, A (Фенилкетонурия III типа) |
6-пирувоилтетрагидроптеринсинтаза (PTPS) |
PTS |
11q22.3-q23.3 |
|
|
233910 |
GCHI |
14q22.1-q22.2 |
||
|
261630 |
Гиперфенилаланинемия, BH4-дефицит, C (Фенилкетонурия II типа) |
дигидроптеридинредуктаза (DHPR) |
QDPR |
4p15.31 |
|
264070 |
птерин-4-альфа-карбиноламиндегидратаза ( PCD ) |
PCBD |
10q22 |
|
|
182125 |
сепиаптеринредуктаза (SR) |
SPR |
1.1.1.153 |
В настоящее время известно несколько форм ВН4 дефицитных ГФА:
- ВН4-дефицитная ГФА (тип А) обусловлена недостаточностью 6-пирувоилтетрагидроптеринсинтазы (PTPS), участвующей в процессе синтеза тетрагидробиоптерина из дигидронеоптерин трифосфата. Заболевание вызвано мутацией структурного гена PTS цитозольной 6-пирувоилтетрагидроптерин синтазы, что приводит к ее недостаточности в печени и эритроцитах. Ген PTS расположен на длинном плече хромосомы 11 в районе q22.3-23.3;
- BH4-дефицитная ГФА (тип В) вследствие недостаточности гуанозин-трифосфат-циклогидролазы I (GTPСH I), кодирующий ген расположен на хромосоме 14q22.2;
-ВН4-дефицитная ГФА (тип С) обусловлена дефицитом дигидроптеридинредуктазы (DHPR), вследствие которого развиваются метаболические блоки на путях превращения фенилаланина в тирозин, а также образования предшественников нейромедиаторов катехоламинового и серотонинового ряда L-дофы и 5-окситриптофана. Заболевание вызвано мутацией структурного гена QDPR цитозольной дигидроптеридинредуктазы. Ген QDPR локализован на хромосоме 4p15.3;
- BH4-дефицитная ГФА (тип D) развивается вследствие недостаточности птерин-4-альфа-карбиноламиндегидратазы (PCВD), которые встречаются реже;
-BH4-дефицитная ГФА вследствие недостаточности сепиаптеринредуктазы (SP).
Тип наследования всех форм ГФА и ФКУ – аутосомно-рецессивный.
ВН4 является кофактором нескольких важных ферментов, в первую очередь фенилаланингидроксилазы, а также тирозингидроксилазы, триптофангидроксилазы и синтазы оксида азота. ВН4-зависимые формы ФКУ имеют сходные клинические проявления с нелеченой классической ФКУ. При этих формах основную роль в патогенезе играет резкая недостаточность нейромедиаторов катехоламинового и серотонинового ряда, поэтому монотерапия диетой не дает положительного результата.
Другие гиперфенилаланинемии могут встречаться при различных физиологических и патологических состояниях.
Транзиторная форма ГФА в период новорожденности - временное повышение уровня ФА в крови ребенка, в большинстве случаев обусловленное незрелостью ферментативных систем новорожденного (например, при глубокой недоношенности или функциональной незрелости). Провоцирующими факторами развития этого состояния у младенца являются преждевременные роды (вследствие чего не успевает повыситься активность парагидроксифенилпируватоксидазы) и чрезмерное употребление белковой пищи матерью. В результате наблюдается повышенная продукция субстрата, ингибирующего собственный фермент, вследствие чего уровень тирозина и фенилаланина в крови увеличивается до патологических значений. Впоследствии биохимические показатели нормализуются. Клинические проявления либо отсутствуют, либо очень незначительны.
Гиперфенилаланинемия, сопровождающая поражение печени различной этиологии (вирусное, медикаментозное, токсическое) и другие наследственные нарушения обмена веществ (лейциноз, классическая галактоземия и др.), имеет вторичный характер.
2. Диагностика
2.1 Жалобы и анамнез
При отсутствии лечения на первом году жизни, обычно в возрасте 2-6 месяцев, родителей беспокоит вялость ребенка, отсутствие интереса к окружающему, иногда повышенная раздражительность, беспокойство, срыгивания, нарушение мышечного тонуса (чаще мышечная гипотония), признаки атопического дерматита, задержка психомоторного развития, иногда судороги. При своевременно назначенном патогенетическом лечении жалобы имеют более легкий характер или отсутствуют.
2.2 Физикальное обследование
При отсутствии лечения обращают на себя внимание фенотипические особенности: гипопигментация кожи, волос, радужной оболочки глаз, своеобразный «мышиный» запах мочи больных, возможно формирование микроцефалии. В психоневрологическом статусе отмечаются задержка статико-моторного и психоречевого развития, симптоматической эпилепсии, а в некоторых случаях гидроцефалии.
Эпилептические приступы встречаются почти у половины нелеченных больных и в некоторых случаях могут служить первым признаком болезни. Обычно отмечаются генерализованные пароксизмы по типу инфантильных спазмов в виде «салаамовых» судорог, «кивков», могут наблюдаться абсансы. Приступы носят упорный характер и плохо поддаются антиконвульсантной терапии. При отсутствии патогенетического лечения болезнь медленно прогрессирует. Умственная отсталость достигает, как правило, глубокой степени, IQ составляет около 20 единиц (норма 85-115 единиц). В психологическом статусе больных отмечают нарушение игровой и предметной деятельности, отсутствие дифференцировки эмоциональных реакций, недостаточность экспрессивной и импрессивной речи. Могут наблюдаться двигательные, психопатоподобные и шизофреноподобные расстройства.
Аналогичные клинические симптомы, которые манифестируют после 2-х месяцев жизни и достигают максимального проявления к 6-ти месяцам жизни имеют BH4-дефицитная ГФА (тип A) вследствие недостаточности 6-пирувоилтетрагидроптеринсинтазы (PTPS), BH4-дефицитная ГФА (тип В) вследствие недостаточности GTP циклогидролазы (GTPSH), BH4-дефицитная ГФА (тип С) вследствие недостаточности дигидроптеридинредуктазы (DHPR), BH4-дефицитная ГФА вследствие недостаточности сепиаптеринредуктазы (Характерно прогрессирующее нарушение психомоторного развития, экстрапирамидные расстройства в виде изменения мышечного тонуса (гипотония мышц туловища и гипертонус мышц конечностей), тремор, атаксия, позднее – нарушения походки, патологические рефлексы, гиперсаливация, нарушение терморегуляции, псевдобульбарные расстройства в виде затруднения глотания, поперхиваний во время приема пищи, микроцефалия, судороги, окулогирные кризы (эпизодическое содружественное отклонение глаз обычно направленное вверх и латерально, редко — вниз или строго латерально), экзема, гипопигментация кожи. У таких детей при рождении нередко отмечается низкая масса тела.
При недостаточности 6-пирувоилтетрагидроптеринсинтетазы (PTPS) различают два фенотипа. Наиболее часто (80%) встречающаяся тяжелая центральная (типичная) форма сопровождается резким дефицитом трансмиттеров и более выраженной тяжестью течения. Вторая, умеренная, периферийная форма сопровождается нормальным содержанием медиаторов, умеренной ГФА и умеренно выраженной клинической симптоматикой.
Для BH4-дефицитной ГФА (тип D) вследствие недостаточности птерин-4-альфа-карбиноламиндегидратазы (PCВD) также характерны специфические изменения мышечного тонуса: постуральная нестабильность, гипокинезия, мышечная дистония (гипертонус конечностей, сниженный тонус мышц туловища).
2.3 Лабораторная диагностика
-
Рекомендуется проведение неонатального скрининга (определение концентрации фенилаланина в сухих пятнах крови) для доклиничской диагностики гиперфенилаланинемии и своевременного начала патогенетической терапии [1,4,11,14,19].
(Сила рекомендации A; уровень убедительности доказательств II)
Комментарии: Все формы ГФА можно диагностировать уже в первые недели или даже дни жизни ребенка, когда клинические проявления еще отсутствуют. Для этого проводят биохимический скрининг новорожденных на наличие гиперфенилаланинемии. Подробная схема проведения неонатального скрининга представлена в приложении Г2, описание правил взятия крови представлено в приложении Г3.
-
Рекомендуется использовать для неонатального скрининга методы флюориметрии или тандемной масс-спектрометрии [1,4,11,14].
(Сила рекомендации A; уровень убедительности доказательств II)
Комментарии: Флюориметрия – количественный биохимический метод определения фенилаланина в крови с помощью современных автоматических флюориметров широко используется для проведения массового автоматизированного скрининга; тандемная масс-спектрометрия – аналитический метод исследования, основанный на определении отношения массы к заряду ионов, образующихся при ионизации исследуемых компонентов пробы, осуществляет качественную и количественную идентификацию аминокислот, позволяет одновременно определять уровень фенилаланина и тирозина, соотношение концентраций позволяет косвенно оценить степень снижения активности фенилаланингидроксилазы.
-
Главным критерием диагностики ГФА рекомендовано считать повышенное содержание фенилаланина в крови.
(Сила рекомендации A; уровень убедительности доказательств II)
Комментарии: Для всех указанных методов уровень ФА в крови человека выше 2,0 мг/дл (120 мкмоль/л) квалифицируют как гиперфенилаланинемию. ГФА с уровнем выше 10,0 мг/дл (600 мкмоль/л) относят к различным формам ФКУ.
-
Рекомендуется проведение дифференциальной диагностики ФКУ и других ГФА (второй этап скрининга) [1,4,11,15,19].
(Сила рекомендации A; уровень убедительности доказательств II)
Комментарии. Второй этап скрининга необходим для выявления ВН4 зависимых форм гиперфенилаланинемии и своевременного назначения патогенетической терапии. В настоящий момент для этого используется определение соотношения фенилаланин/тирозин (косвенно отражает наличие дефицита фермента РАН) и ДНК диагностика [4,7,14,17].
-
При отсутствии данных неонатального скрининга диагностику заболевания рекомендовано осуществлять на основании совокупности генеалогического анамнеза, результатов клинического и биохимического обследования [4,7,14,15,20].
(Сила рекомендации С; уровень убедительности доказательств II)
Комментарии: В этих случаях главным для установления диагноза остается биохимический критерий - высокое содержание фенилаланина в сыворотке крови при отсутствии патогенетического лечения. В дальнейшем показано проведение молекулярной диагностики.
-
Рекомендуется ДНК диагностика с целью выявления мутаций в генах РАН и РТР.
(Сила рекомендации A; уровень убедительности доказательств II)
Комментарии. ДНК диагностика проводится с целью дифференциальной диагностики и для определения характера мутаций, что в дальнейшем помогает определить контингет пациентов для проведения теста на чувствительность к лечению сапроптерином Существующие наборы позволяют определять частые мутации в гене РАН, имеющиеся у 80% больных ФКУ. При отсутствии исследуемых мутаций у пациента рекомендуется проведение секвенирования гена РАН. Также проводится ДНК диагностика мутаций и секвенирование генов (PTPS, DHPR, PCD и др.), кодирующих известные ферменты различных стадий метаболизма тетрагидробиоптерина.
В настоящее время известно более 800 мутаций в гене РАН, спектр и распространенность которых имеет этнические особенности. Для европеоидной расы мажорной мутацией в гене РАН является мутация R408W, в то время как в Японии и Китае данная мутация не найдена. Во многих европейских популяциях с относительно высокой частотой регистрируются следующие мутации: IVS12nt1, R261Q, R252W, R158Q, P281L, IVS10nt546, I65T.
Большинство генетических изменений гена РАН – это миссенс-мутации во всех 13 экзонах гена или нетранслируемых фланкирующих участках гена, составляющие 59% всех вариантов. Также обнаружены мутации сплайсинга, нонсенс-мутации, мутации сдвига рамки считывания, более крупные делеции и инсерции. Разные мутации влияют на работу фермента РАН в различной степени – этим может объясняться большое разнообразие показателя ФА в крови больных ФКУ. Большое количество мутаций гена РАН (например, R408W) приводят к резидуальной активности фермента ФАГ. При других мутациях (E390G, Y414C,A300) толерантность к фенилалалнину выше и клиническая картина ФКУ менее выражена. Таким образом, по современным данным (Hentz et al., 2013) результаты генотипирования при ФКУ потенциально обладают предиктивным значением.
Следует отметить, что при определенных мутациях в гене РАН, обусловливающих остаточную активность фермента ФАГ, введение кофактора BH4 или его синтетических аналогов в терапию приводит к повышению или восстановлению активности фенилаланингидроксилазы, что позволяет расширить диету с увеличением в рационе квоты натурального белка.
2.4 Инструментальная диагностика
-
Рекомендуется электроэнцефалография для выявления патернов гипсаритмии (даже при отсутствии клинических судорожных приступов), единичных и множественных спайк- и полиспайкразрядов, других изменений [4,13]
(Сила рекомендации С; уровень убедительности доказательств II)
-
Рекомендуется магнитнорезонансная томография с целью выявления очагов перивентрикулярной лейкопатии, кортикальной атрофии и других изменений у пациентов старше 12 лет [4,21,24].
(Сила рекомендации С; уровень убедительности доказательств II)
-
Рекомендуется ультразвуковое исследование брюшной полости и почек для диагностики дискинезии желчных путей, диффузных изменений печени и поджелудочной железы, мочекаменной болезни [1,2.4].
(Сила рекомендации С; уровень убедительности доказательств II)
-
Рекомендуется эзофагогастрофиброскопия для диагностики поражения слизистой желудка ( по показаниям) [1,2,4,24].
(Сила рекомендации С; уровень убедительности доказательств II)
2.5 Иная диагностика (консультативная помощь)
-
Рекомендуется психолого-педагогическое консультирование и логопедическое тестирование [1,4,6,13].
(Сила рекомендации С; уровень убедительности доказательств II)
Комментарии: Тестирование проводится с целью определения уровня интеллектуального и речевого развития, возможностей социальной адаптации и составления индивидуального плана психолого-педагогического сопровождения.
-
Рекомендуются по показаниям консультации специалистов (невропатолог, гастроэнтеролог, офтальмолог, аллерголог и др.) [1,2,9,].
(Сила рекомендации С; уровень убедительности доказательств II)
2.6. Дифференциальная диагностика
-
Рекомендовано проводить дифференциальную диагностику с целью выявления ВН4-зависимых форм ГФА и для определения потенциально чувствительных больных с РАН зависимой ГФА (фенилкетонурией).
(Сила рекомендации С; уровень убедительности доказательств II)
-
Рекомендовано после подтверждения у новорожденного РАН-зависимой гиперфенилаланинемии провести тест на потенциальную чувствительность к сапроптерина дигидрохлориду, после чего назначают лечение [16,18,20].
(Сила рекомендации С; уровень убедительности доказательств II)
Комментарий: Процедура проведения тестирования описана в приложении Г3.
Уровень фенилаланина в крови до начала тестирования должен быть ?450 мкмоль/л.
Дополнительную информацию для дифференциальной диагностики можно получить при исследовании птеринов мочи, сыворотки крови и спинномозговой жидкости (в РФ не проводится) (таблица 3).
Таблица 3 - Диагностические показатели фенилананина и птеринов в крови, моче и спинномозговой жидкости (N. Blau et al. 2014) *.
|
фермент |
Phe (К) |
Neo (К,М) |
Bio (К,М) |
Pri (К,М) |
Neo (СМЖ) |
Bio (СМЖ)
|
SHIAA (СМЖ) |
HVA (СМЖ) |
SMTHF (СМЖ) |
активность DHPR (К) |
|
AdGTPSH |
n |
n |
n |
n |
? |
? |
n-? |
? |
n |
n |
|
ArGTPSH |
? |
? |
? |
n |
? |
? |
? |
? |
n |
n |
|
PTPS |
? |
? |
? |
n |
? |
? |
? |
? |
n |
n |
|
SR |
n |
n |
n |
n |
n |
?** |
? |
? |
n |
n |
|
PCD |
? |
? |
n-? |
? |
n |
n |
n |
n |
n |
n |
|
DHPR |
? |
n |
? |
n |
n |
? |
? |
? |
n-? |
? |
 показатели
показатели*- AdGTPSH-аутосомно-доминантный дефицит гуанозин-трифосфат-циклогидролазы I; ArGTPSH-аутосомно-рецессивный дефицит гуанозин-трифосфат-циклогидролазы I, PTPS-6-пирувоилтетрагидроптеринсинтаза, SR-сепиаптеринредуктаза, PCD-птерин-4-альфа-карбиноламиндегидратаза, DHPR-дигидроптеридинредуктаза, Phe-фенилаланин, Neo-неоптерин, Bio-биоптерин, Pri-примаптерин,. SHIAA-S-гидроксииндолуксусная кислота, HVA- гомованилиновая кислота, SMTHF – 5-метитетрагидрофолат, К-кровь, М-моча. СМЖ-спиномозговая жидкость.
3. Лечение
Основная цель лечения — снизить ФА крови, повысить толерантность (переносимость) ФА, получаемого с натуральной пищей и таким образом избежать тяжелой неврологической симптоматики и улучшить качество жизни.
Подходы к терапии при различной тяжести течения гиперфенилаланинемии:
Легкая гиперфенилаланинемия (ГФА) требует наблюдения и проведения дифференциальной диагностики. Строгого диетического лечения при этой форме ГФА, как правило, не назначают, хотя в последние годы рекомендуют начинать лечение при уровне ФА в крови ?360 мкмоль/л (N Blau 2014). Дети с легкой формой ГФА должны находиться под систематическим наблюдением врача в течение первого года жизни с контролем уровня ФА крови, проведением необходимых диагностических мероприятий с целью исключения птерин-зависимых форм ГФА и выбора дальнейшей тактики лечения.
Умеренная (мягкая, средняя) ФКУ подразумевает сохранение частичной активности фермента фенилаланингидроксилазы, требует назначения гипофенилаланиновой диеты, а также проведения теста на чувствительность к терапии синтетическим аналогом тетрагидробиоптерина.
Классическая (тяжелая) ФКУ обусловлена минимальной активностью фермента, требует назначения строгой гипофенилаланиновой диеты, а также проведения теста на чувствительность к терапии синтетическим аналогом тетрагидробиоптерина.
Патогенетически обоснованной терапией для больных с ГФА-обусловленной недостаточностью тетрагидробиоптерина является назначение синтетического аналога ВН4 – сапроптерина дигидрохлорида, который используется в комплексе с диетотерапией или без нее (в зависимости от формы болезни) и симптоматической медикаментозной терапией.
3.1 Консервативное лечение
-
Рекомендуется патогенетически обоснованная для РАН-зависимой ФКУ и ГФА гипофенилаланиновая диета [1,3,12,24].
(Сила рекомендации С; уровень убедительности доказательств II)
Комментарии: Диетотерапия, основанная на резком ограничении фенилаланина в рационе больных детей за счет исключения высокобелковых продуктов, должна быть начата не позднее первых недель жизни ребенка с целью достижения максимальной эффективности лечения. Недостающее количество белка восполняется за счет специализированных лечебных продуктов, частично или полностью лишенных фенилаланина.
Согласно современным данным, диетическое лечение назначают при уровне ФА на скрининге ?360 мкмоль/л (?6 мг/дл).
Из рациона питания больного ФКУ исключают продукты с высоким содержанием белка (соответственно и фенилаланина): мясо, мясопродукты, рыбу, рыбопродукты, творог, яйцо, бобовые, орехи, шоколад и др. Допустимые в диете натуральные продукты, такие как женское молоко, детские молочные смеси (для детей в возрасте до 1 года), овощи, фрукты и некоторые другие продукты с низким содержанием белка вводят в соответствии с подсчетом содержащегося в них фенилаланина.
При расчетах питания рекомендуется ориентироваться на нормы физиологической потребности в основных нутриентах для больных детей различных возрастных групп в соответствии с МР 2.3.1.2432-08 (таблицы 4, 5), допускается уменьшение количества суточного белка (не более 10%) в зависимости от толерантности больного к пище и к фенилаланину, а также от состояния нутритивного статуса.
Таблица 4 - Среднесуточные нормы потребностей в основных пищевых веществах и энергии для детей первого года жизни (на кг массы тела) [12].
|
Возраст (мес.) |
Энергия, ккал/ кг |
Белок, г/кг |
Жиры, г/кг |
Углеводы, г/кг |
|
0-3 |
115 |
2,2 |
6,5 |
13 |
|
4-6 |
115 |
2,6 |
6,0 |
13 |
|
7-12 |
110 |
2,9 |
5,5 |
13 |
Таблица 5 - Нормы физиологической потребности в основных пищевых веществах и энергии для детей старше года [12].
|
Возраст |
Энергия, ккал |
Белок, г/день* |
Жиры, г/день |
Углеводы, г/день |
|
от 1 года до 2 лет |
1200 |
36 (28) |
40 |
174 |
|
от 2 лет до 3 лет |
1400 |
42 (33) |
47 |
203 |
|
от 3 лет до 7 лет |
1800 |
54 (46) |
60 |
261 |
|
от 7 лет до 11 лет |
2100 |
63 (54) |
70 |
305 |
|
от 11 лет до 14 лет мальчики |
2500 |
75 (64) |
83 |
363 |
|
от 11 лет до 14 лет девочки |
2300 |
69 (59) |
77 |
334 |
|
от 14 лет до 18 лет юноши |
2900 |
87 (74) |
97 |
421 |
|
от 14 лет до 18 лет девушки |
2500 |
76 (64) |
83 |
363 |
* В скобках указано ориентировочное потребление белка за счёт специализированной смеси без фенилаланина.
Белок за счет естественных продуктов в диете рассчитывается, исходя из допустимых суточных количеств ФА с учетом, что 1 г белка содержит приблизительно 50 мг ФА. В зависимости от переносимости пищевого фенилаланина допустимое и безопасное количество ФА в сутки составляет от 90 до 35 мг/кг массы тела для детей первого года жизни. В питании детей старше года допустимое количество ФА постепенно снижается от 35 до 10 мг/кг массы тела ребенка (таблица 6).
Таблица 6 - Допустимое количество фенилаланина в питании детей с ФКУ в зависимости от возраста
|
Возраст детей |
Количество фенилаланина (мг/кг массы тела в сутки) |
|
от 0 до 2 месяцев |
90-60 |
|
2-6 месяцев |
55-45 |
|
6-12 месяцев |
40-35 |
|
1-3 |
35-25 |
|
3-7 |
25-20 |
|
7 и старше |
20-10 |
Недостающее количество белка восполняется за счет специализированных лечебных продуктов - смесей аминокислот без фенилаланина и низкобелковых продуктов питания. Аминокислотные смеси различаются по содержанию белка (от 13 г до 77,5 г на 100 г сухого продукта) и других питательных веществ (углеводы, жиры, витамины, микро- и макроэлементы). Все смеси в своем составе не содержат фенилаланин. Аминокислотные смеси с содержанием 13-15 г белка в 100 г сухой смеси предназначены для детей первого года жизни. Детям более старшего возраста назначаются смеси с более высоким содержанием белка (приложение Г4).
На первом году жизни необходим ежедневный подсчет количества фенилаланина, получаемого с пищей, употребляемой пациентом, учет белков, жиров, углеводов, энергии в суточном рационе.
Расчет суточной дозы специализированного продукта производится по формуле: (Ps-Pn) х 100
P
Ps - суточное количество белка,
Pn - белок естественных продуктов,
P - количество белка в 100 г сухого специализированного продукта
Пример расчета питания ребенку с ФКУ
Ребенок 3 лет, масса тела 14,5 кг
- Общее суточное количество белка в рационе больного (см. табл.5): 54,0г
- Допустимое суточное количество фенилаланина (см. табл.6):
25 х 14,5 = 363 мг
- Допустимое количество белка естественных продуктов (1г белка содержит 50 мг фенилаланина):
З6З:50 = 7,3 г
- Количество белка за счет специализированного продукта на основе смеси L-аминокислот без фенилаланина:
54,0 - 7,3 = 46,7 г
- Суточное количество аминокислотной смеси (100 г содержит 20 г белка):
46,7 x 100:20 = 233 г
- Рекомендуемое суточное количество жира в рационе (см. табл.5): 60 г.
- Рекомендуемое суточное количество углеводов (см. табл.5): 261 г.
При организации диетотерапии необходимо учитывать клиническую форму заболевания, уровень ФА в крови, возраст ребенка; нутритивный статус (физическое развитие), толерантность ребенка к пищевому ФА, количество ФА и натурального белка, получаемого с пищей, количество основных пищевых веществ и энергии в лечебном рационе.
При назначении диеты важен индивидуальный и дифференцированный подход к использованию специализированных и натуральных продуктов соответственно возрасту ребенка.
Для больных ФКУ независимо от возраста, сохраняется запрет на продукты, наиболее богатые ФА, такие как мясо, рыба и изделия из них. Творог, твёрдые сыры, бобовые, куриные яйца, орехи могут в ограниченном количестве входить в рацион пациентов старшего возраста с учетом толерантности к ФА. Не рекомендуется употребление пациентами с ФКУ продуктов «fast food», газированных напитков с подсластителями (аспартам или пищевая добавка Е951), содержащими ФА.
Тактика диетотерапии при сопутствующих заболеваниях (выраженная гипертермия, интоксикация, различные диспепсические явления), а также при отказе от приема аминокислотной смеси заключается в кратковременном (на 2-3 дня) прекращении диетотерапии с заменой лечебных продуктов на натуральные с невысоким содержанием белка. После стихания острого периода болезни в рацион ребенка вновь вводится специализированный продукт, но за более короткий период, чем в начале лечения. Если ребенок с ФКУ не отказывается от пищи во время болезни, то лечение сопутствующих соматических проводится по общепринятой схеме и не требует прекращения диетотерапии.
-
Рекомендуется ( по показаниям) медикаментозная терапия сапроптерином ФКУ, обусловленной дефектами обмена фенилалангидроксидазы (легкие и умеренные формы) после проведения теста и подтверждения чувствительности к сапроптерину [5,7,141524].
(Сила рекомендации С; уровень убедительности доказательств II)
Комментарии: Синтетический аналог ВН4 – сапроптерина дигидрохлорид является патогенетическим методом лечения для ВН4-дефицитных форм ГФА и вспомогательным методом лечения чувствительных к ВН4 терапии форм классической ФКУ.
Тестирование потенциальной чувствительности к лечению и лечение сапроптерином проводит и контролирует врач, который осуществляет лечение и наблюдение пациентов с ФКУ ( Приложение Г5).
Ответ на лечение препаратом сапроптерина дигидрохлоридом оценивается по степени снижения концентрации фенилаланина в крови больного при соблюдении стабильной гипофенилаланиновой диеты. Пациент считается чувствительным, если разница уровня ФА, полученного по окончании периода оценки ответа на лечение, и исходного уровня ФА перед началом приема препарата составляет 30% и более.
При необходимости длительная медикаментозная терапия у больных ФКУ, отвечающих на лечение сапроптерина дигидрохлоридом снижением уровня ФА в крови, проводится в комбинации с диетой при использовании аминокислотных смесей, количество которых определяет врач.
3.4 Иное лечение
Для ВН4-дефицитных форм ГФА патогенетическим методом лечения рекомендуется применение синтетического аналога ВН4 – сапроптерина дигидрохлоридж (А16АХ) в сочетании с гипофенилаланиновой диетой.
Медикаментозная терапия при ВН4-дефицитных состояниях с ГФА и без ГФА: начальная доза сапроптерина дигидрохлорида у больных с недостаточностью BH4 составляет от 2 до 5 мг/кг массы тела при приеме 1 раз в день. Доза может быть увеличена до 20 мг/кг массы тела в день. Для достижения оптимального терапевтического эффекта суточная доза препарата может быть разделена на 2 или 3 приема в течение дня.
В комплекс лечения также входят препараты леводопыж,вк (10-15 мг/кг/сут) в сочетании с карбидопойж,вк (Код АТХ: N04BA)в дозировке 1-1,5 мг/кг/сут, 5-гидрокситриптофан (10 мг/кг/сут) (препарат в Российской Федерации в настоящее время не зарегистрирован), 5-формилтетрагидрофолат (кальция фолинатж,вк - Код АТХ: V03AF) в средней дозе 25 мг/сут, в некоторых случаях диета с ограничением ФА и фолиевая кислотаж,вк (Код АТХ: B03BB) (таблица 7).
Таблица 7 - Схема терапии при различных ВН4 дефицитных состояниях (с ГФА и без ГФА)*
|
Ферменты |
BH4 |
L-допа |
Карбидопа |
5-гидрокси триптофан** |
Диета с низким ФА |
Фолиевая кислота |
|
GTPCH |
+ |
+ |
+ |
+ |
- |
- |
|
DRD/аутосомно-доминантный тип GTPCH |
- |
+ |
+ |
- |
- |
- |
|
PTPS умеренная форма |
+ |
- |
- |
- |
- |
- |
|
PTPS тяжелая форма |
+ |
+ |
+ |
+ |
- |
- |
|
DHPR |
- |
+ |
+ |
+ |
+ |
+ |
|
SR |
- |
+ |
+ |
+ |
- |
- |
|
PCBD |
+ |
+ |
+ |
+ |
- |
- |
 Средства терапии
Средства терапии*-таблица составлена по данным N.Blau 2014.
**- препарат не зарегистрирован в Российской Федерациии
4. Реабилитация
Реабилитационные мероприятия направлены на улучшение когнитивных и речевых функций, психоэмоционального состояния, социальной адаптации.
5. Профилактика и диспансерное наблюдение
5.1 Профилактика
Профилактика фенилкетонурии и гиперфенилаланинемии проводится в нескольких направлениях:
- проспективное медико-генетическое консультирование пар, планирующих беременность с рекомендацией обследования на гетерозиготное носительство частых мутаций в гене РАН. При выявлении ФКУ в семье – обследование родственников для уточнения гетерозиготного носительства мутации;
- в семье, где имеется ребенок с ФКУ, при последующей беременности проведение пренатальной диагностики для определения наличия патологии у плода;
- проведение неонатального скрининга с охватом 100% новорожденных, позволяющего рано выявить заболевание, своевременно начать лечение и избежать тяжелых проявлений патологии;
-профилактика рождения детей с синдромом «материнской фенилкетонурии» от женщин, больных ФКУ, путем организации психологической помощи девочкам-подросткам по вопросам необходимости соблюдения строгой гипофенилаланиновой диеты в пубертатный период, а также консультативной помощи по вопросам планирования семьи и беременности.
5.2 Диспансерное наблюдение
Заболевание диагностируется в результате неонатального скрининга, выявленные больные находятся на диспансерном учете в медико-генетических центрах (консультациях), где назначается лечение, включающее диетотерапию и осуществляется контроль за адекватностью его проведения.
- Рекомендуется следующая схема контроля за содержанием ФА в крови у больных ФКУ:
-в возрасте до 3-х месяцев - 1 раз в неделю (до получения стабильных результатов) и далее 1 раз в 10 дней,
-с 1 года до 6 лет – не менее 2 раз в месяц,
-с 7 до 12 лет - не менее 1 раза в месяц,
-после 12 лет - 1 раз в 2 месяца,
Терапевтический диапазон уровня ФА в сыворотке крови может быть расширен в зависимости от возраста и ослабления диетических ограничений (таблица 8).
Таблица 8 - Рекомендуемый уровень ФА в сыворотке крови у больных ФКУ, находящихся на лечении
|
Возраст и периоды жизни пациентов |
Уровень ФА |
|
|
мкмоль/л |
мг/дл |
|
|
0-6 лет |
120-360 |
2-6 |
|
7-9 лет |
120-360 |
2-6 |
|
10-12 лет |
120-360 |
2-6 |
|
13-15 лет |
120-600 |
2-10 |
|
16-18 лет |
120-900 |
2-15 |
На фоне лечения необходимо проводить контроль за нутритивным статусом больного, физическим и интеллектуальным развитием. Большинство детей с ФКУ находятся на этапе амбулаторно-поликлинического наблюдения, где осуществляется контроль за состоянием их здоровья с привлечением врачей специалистов, использованием функциональных методов исследования (УЗИ, ЭЭГ, МРТ), а также контроль клинико-лабораторных показателей (общие анализы крови и мочи, общий белок и его фракции, по показаниям липидный профиль, глюкоза, ферритин, креатинин, сывороточное железо и др.), 1 раз в году рекомендуется исследовать аминокислотный спектр крови. Общий анализ крови рекомендуется делать не реже 1 раза в 6 месяцев, биохимический – 1 раз в год. Консультация психолога для больных ФКУ подросткового возраста является обязательной и должна проходить ежегодно. Контроль фосфорно-кальциевого обмена (кальций, фосфор, остеокальцин, паратгормон и др.) должен проводиться с 1 года жизни. Пациентам старше 13 лет рекомендуется проведение денситометрии 1 раз в год.
Девочкам с ФКУ рекомендуется поддерживать содержание ФА в крови на уровне до 4 мг/дл (240 мкмоль/л) независимо от возраста с целью профилактики в дальнейшем синдрома «материнской фенилкетонурии» у будущего потомства [8,22,23].
6. Дополнительная информация, влияющая на течение и исход заболевания
6.1 Исходы и прогноз
Прогноз заболевания зависит от своевременной диагностики, максимально раннего назначения диетотерапии и адекватного ее выполнения и контроля. Большое значение имеет приверженность лечению всех членов семьи больного, а в дальнейшем - и самого пациента, для этого необходимо организовать психолого-педагогическую поддержку, которая должна начинаться с момента рождения больного ребенка.
В случае несоблюдения рекомендаций по диетотерапии и недостаточном контроле за уровнем ФА в крови могут иметь место такие отдаленные последствия, как сниженный коэффициент интеллекта, нарушения речи и памяти, проблемы с концентрацией внимания, расстройства поведения.
В случае, если пациенты не принимают специализированные аминокислотные смеси без фенилаланина, а находятся на диете с резким ограничением высокобелковых продуктов, возможно развитие симптомов хронической недостаточности питания, нутритивного дефицита витаминов, макро- и микроэлементов и других эссенциальных факторов.
Для классической ФКУ, выявленной в первые недели жизни ребёнка, при соблюдении рекомендаций врачей по лечению, прогноз по заболеванию благоприятный. Дети посещают массовые детские и образовательные учреждения, занимаются в дополнительных кружках, в дальнейшем поступают в высшие учебные заведения.
Критерии оценки качества медицинской помощи
Таблица 1 - Организационно-технические условия оказания медицинской помощи.
|
Вид медицинской помощи |
специализированная, в том числе высокотехнологичная, медицинская помощь |
|
Возрастная группа |
дети |
|
Условия оказания медицинской помощи |
стационарно, в дневном стационаре |
|
Форма оказания медицинской помощи |
плановая |
Таблица 2 - Критерии оценки качества медицинской помощи
Список литературы
-
Баранов А.А., Боровик Т.Э., Ладодо К.С., Бушуева Т.В., Маслова О.И., Кузенкова Л.М., Студеникин В.М., Звонкова Н.Г. и др. Специализированные продукты лечебного питания для детей с фенилкетонурией. Методическое письмо. 3-е издание. Москва. 2012.84с
-
Боровик Т.Э., Ладодо К.С., Грибакин С.Г., Скворцова В.А., Бушуева Т.В., Вознесенская Т.С., Заплатников А.Л., Захарова И.Н., Звонкова Н.Г., Картамышева Н.Н., Коровина Н.А., Кураева Т.Л., Кутафина Е.К., Мазанкова Л.Н., Макарова С.Г., Мухина Ю.Г., Нетребенко О.К., Потапов А.С., Рославцева Е.А., Рыбакова Е.П. и др. Клиническая диетология детского возраста. Руководство для врачей // Под редакцией Т.Э. Боровик, К.С. Ладодо. Москва, 2008.
-
Боровик Т.Э., Ладодо К.С., Бушуева Т.В. и др. Диетотерапия при классической фенилкетонурии: критерии выбора специализированных продуктов без фенилаланина Вопросы современной педиатрии 2013; 12 (5): 40–48).
-
Бушуева Т.В. Современный взгляд на проблему фенилкетонурии у детей: диагностика, клиника, лечение. Вопросы современной педиатрии 2010, том 9, №11, с. 57-162.
-
Бушуева Т.В., Кузенкова Л.М., Боровик Т.Э., Назаренко Л.П. и др. Открытое несравнительное клиническое исследование III фазы по оценке частоты ответа и безопасности сапроптерина у пациентов с фенилкетонурией и гиперфенилаланинемией. Вестник Российской академии медицинских наук.-2014-№ 7–8 стр. 69-77.
-
Голихина Т.А., Люманова Э.Р. Психологический статус личности детей с фенилкетонурией, получающих диетотерапию с раннего возраста // Кубанский научный медицинский вестник.- 2011.- №8(122).– С.50-53.
-
Гундорова П, Степанова А.А., Щагина О.А., Поляков А.В. Результаты использования новых медицинских технологий «Детекция основных точковых мутаций гена РАН методом мультиплексной лигазной реакции» и «Детекция десяти дополнительных точковых мутаций гена РАН методом мультиплексной лигазной реакции» в ДНК диагностике фенилкетонурии. Медицинская генетика.-2-16-Т.15-№2(164)-с.29-36.
-
Денисенкова Е.В. Кузнецова Л.И. Влияние материнской фенилкетонурии на исход беременности и родов. Вопросы детской диетологии 2009.т.7, №3, стр.55-59.
-
Ладодо К.С., Рыбакова Е.П., Соломадина Л.В. Специализированное лечебное питание для детей с фенилкетонурией. Руководство по фармакотерапии в педиатрии и детской хирургии. Клиническая генетика. Под ред. Царегородцева А.Д., Таболина В.А., Москва, 2002, с.132-138.
-
Матулевич, С.А. Компьютеризация и программное обеспечение неонатального скрининга на наследственные болезни обмена / С.А. Матулевич // Медицинская генетика. - 2009. –Т.8, №3 (81). - С.35-38.
-
Матулевич С.А., Голихина Т.А. Глава 32. Неонатальный скрининг на наследственные болезни (с.853-887). / Наследственные болезни: национальное руководство / под ред. акад. РАМН Н.П. Бочкова, акад. РАМН Е.К.Гинтера, акад. РАМН В.П.Пузырева. – М.:ГЭОТАР-Медиа. 2012.-936 с.
-
MP 2.3.1.2432-08 "Нормы физиологических потребностей в энергии и пищевых веществах для различных групп населения Российской Федерации" (утв. Главным государственным санитарным врачом РФ 18 декабря 2008 г.).
-
Bushueva T.V., Vinyarskaya I.V., Chernikov V.V., Borovik T.E., Kuzenkova L.M. Quality of life in russian children with phenylketonuria
Journal inherited metabolic disease Vol.37.Suppl.1 September 2014 S60-61.
-
Blau N, Burton BK, Thony B., F J van Spronsen, S.Waisbren. Phenylketonuria and BH4 Deficiencies. UNI-MED Verlag AG. Bremen-London-Boston. 79p.
-
Blau N, Hennermann JB, Langenbeck U, Lichter-Konecki U. Diagnosis,classification, and genetics of phenylketonuria and etrahydrobiopterin (BH4) deficiencies. Mol Genet Metab 2011;104:S2–S9.
-
Burton BK, Grange DK, Milanowski A, et al. The response of patients with phenylketonuria and elevated serum phenylalanine to treatment with oral sapropterin dihydrochloride (6R-tetrahydrobiopterin): a phase II, multicentre, open-label, screening study. J Inherit Metab Dis 2007;30: 700–707.
-
Chace DH, Millington DS, Terada N, Kahler SG, Roe CR, Hofman LF. Rapid diagnosis of phenylketonuria by quantitative nalysis or phenylalanine and tyrosine in neonatal blood spots by tandem mass spectrometry. Clin Chem 1993;39:66–71.
-
Cunningham A, Bausell H, Brown M, et al. Recommendations for the use of sapropterin in phenylketonuria. Mol Genet Metab 2012;106:269–276.
-
Howell RR. National Institutes of Health Consensus Development Conference Statement: phenylketonuria: screening and management, October 16–18, 2000. Pediatrics 2001;108(4):972–982.
-
Gordon P, Thomas JA, Suter R, Jurecki E. Evolving patient selection and clinical benefit criteria for sapropterin dihydrochloride (Kuvan®) treatment of PKU patients. Mol Genet Metab 2012;105:672–676.
-
Levy HL, Milanowski A, Chakrapani A, et al.; Sapropterin Research Group.Efficacy of sapropterin dihydrochloride tetrahydrobiopterin, 6R-BH4) for reduction of phenylalanine concentration in patients with phenylketonuria: a phase III randomised placebo-controlled study. Lancet 2007;370: 504–510.
-
Lee PJ, Ridout D, Walter JH, Cockburn F. Maternal phenylketonuria: report from the United Kingdom Registry 1978-97. Arch Dis Child 2005;90:143–146
-
Ng TW, Rae A, Wright H, Gurry D, Wray J. Maternal phenylketonuria in Western Australia: pregnancy outcomes and developmental outcomes in offspring. J Paediatr Child Health 2003;39:358–363.
-
Singh RH, Rohr F, Frazier D, et al. Recommendations for the nutrition management of phenylalanine hydroxylase deficiency. Genet Med, e-pub ahead of print 2 January 2014.
-
Anjema K, van Rijn M, Hofstede FC, Bosch AM, Hollak CE, Rubio-Gozalbo E, de Vries MC, Janssen MC, Boelen CC, Burgerhof JG, Blau N, Heiner-Fokkema MR, van Spronsen FJ. Tetrahydrobiopterin responsiveness in phenylketonuria: prediction with the 48-hour loading test and genotype. Orphanet J Rare Dis. 2013 Jul 10;8:103.
-
Burton BK1, Grange DK, Milanowski A, Vockley G, Feillet F, Crombez EA, Abadie V, Harding CO, Cederbaum S, Dobbelaere D, Smith A, Dorenbaum A. The response of patients with phenylketonuria and elevated serum phenylalanine to treatment with oral sapropterin dihydrochloride (6R-tetrahydrobiopterin): a phase II, multicentre, open-label, screening study. J Inherit Metab Dis. 2007 Oct;30(5):700-7.
Приложение А1. Состав рабочей группы
-
Баранов А.А. – академик РАН, профессор, д.м.н., Председатель Исполкома Союза педиатров России.
-
Намазова-Баранова Л.С. - академик РАН, проф., д.м.н. заместитель Председателя Исполкома Союза педиатров России.
-
Боровик Т.Э. – д.м.н., проф., член Исполкома Союза педиатров России.
-
Бушуева Т.В. – д.м.н., член Союза педиатров России.
-
Бакулина Е.Г. –к.м.н., член Ассоциации медицинских генетиков
-
Голихина Т.А. – к.м.н., член Ассоциации медицинских генетиков
-
Гундарова П. – аспирант
-
Денисенкова Е.В. – к.м.н., член Ассоциации медицинских генетиков
-
Журкова Н.В. – к.м.н., член Союза педиатров России
-
Захарова Е.Ю. – д.м.н., проф., член Ассоциации медицинских генетиков
-
Звонкова Н.Г. - к.м.н., член Союза педиатров России.
-
Кузенкова Л.М. - д.м.н., проф., член Исполкома Союза педиатров России.
-
Куцев С.И. - д.м.н., проф.. член-корр. РАН.
-
Ладодо К.С. – д.м.н., проф., член Союза педиатров России
-
Лязина Л.В. – к.м.н., член Ассоциации медицинских генетиков
-
Матулевич С.А. –д.м.н., член Ассоциации медицинских генетиков
-
Назаренко Л.П. – д.м.н., проф., член Ассоциации медицинских генетиков
-
Николаева Е.А. - д.м.н., проф., член Ассоциации медицинских генетиков
-
Николаева Е.Б. –д.м.н., член Союза педиатров
-
Никитина Н.В.- к.м.н., член Ассоциации медицинских генетиков
-
Новиков П.В. - д.м.н., проф., член Ассоциации медицинских генетиков
-
Поляков А.В. – д.б.н., проф., член Ассоциации медицинских генетиков
-
Пушков А.А. - к.б.н., член Союза педиатров России.
-
Романенко О.П. – д.м.н., проф., член Ассоциации медицинских генетиков
-
Савостьянов К.В. - к.б.н., член Союза педиатров России.
Авторы подтверждают отсутствие финансовой поддержки/конфликта интересов, который необходимо обнародовать.
Приложение А2. Методология разработки клинических рекомендаций
Целевая аудитория данных клинических рекомендаций:
-
врачи педиатры
-
врачи общей семейной практики (семейная медицина)
-
генетики
-
диетологи
-
невропатологи
-
медицинские психологи
-
дефектологи
Таблица П1 – Уровни убедительности доказательств
|
Уровни убедительности доказательств |
Источник доказательств |
|
I (1) |
Проспективные рандомизированные контролируемые исследования Достаточное количество исследований с достаточной мощностью, с участием большого количества пациентов и получением большого количества данных Крупные мета-анализы Как минимум одно хорошо организованное рандомизированное контролируемое исследование Репрезентативная выборка пациентов |
|
II (2) |
Проспективные с рандомизацией или без исследования с ограниченным количеством данных Несколько исследований с небольшим количеством пациентов Хорошо организованное проспективное исследование когорты Мета-анализы ограничены, но проведены на хорошем уровне Результаты не презентативны в отношении целевой популяции Хорошо организованные исследования «случай-контроль» |
|
III (3) |
Нерандомизированные контролируемые исследования Исследования с недостаточным контролем Рандомизированные клинические исследования с как минимум 1 значительной или как минимум 3 незначительными методологическими ошибками Ретроспективные или наблюдательные исследования Серия клинических наблюдений Противоречивые данные, не позволяющие сформировать окончательную рекомендацию |
|
IV (4) |
Мнение эксперта/данные из отчета экспертной комиссии, экспериментально подтвержденные и теоретически обоснованные |
Таблица П2 – Сила рекомендаций
|
Сила рекомендаций |
Описание |
Расшифровка |
|
A |
Рекомендация основана на высоком уровне доказательности (как минимум 1 убедительная публикация I уровня доказательности, показывающая значительное превосходство пользы над риском) |
Метод/терапия первой линии; либо в сочетании со стандартной методикой/терапией |
|
B |
Рекомендация основана на среднем уровне доказательности (как минимум 1 убедительная публикация II уровня доказательности, показывающая значительное превосходство пользы над риском) |
Метод/терапия второй линии; либо при отказе, противопоказании, или неэффективности стандартной методики/терапии. Рекомендуется мониторирование побочных явлений |
|
C |
Рекомендация основана на слабом уровне доказательности (но как минимум 1 убедительная публикация III уровня доказательности, показывающая значительное превосходство пользы над риском) или нет убедительных данных ни о пользе, ни о риске) |
Нет возражений против данного метода/терапии или нет возражений против продолжения данного метода/терапии Рекомендовано при отказе, противопоказании, или неэффективности стандартной методики/терапии, при условии отсутствия побочных эффектов |
|
D |
Отсутствие убедительных публикаций I, II или III уровня доказательности, показывающих значительное превосходство пользы над риском, либо убедительные публикации I, II или III уровня доказательности, показывающие значительное превосходство риска над пользой |
Не рекомендовано |
Приложение А3. Связанные документы
-
Приказ Минздравсоцразвития РФ № 185 от 22.03.2006 года «О массовом обследовании новорожденных детей на наследственные заболевания»,
-
Приказ Министерства здравоохранения и социального развития РФ от 16 апреля 2012 г. N 366н "Об утверждении Порядка оказания педиатрической помощи"
-
Приказ Министерства здравоохранения РФ "Об утверждении Порядка оказания медицинской помощи больным с врожденными и (или) наследственными заболеваниями"от 15 ноября 2012 г. N 917н
-
Постановление Правительства Российской Федерации от 9 апреля 2015 года №333 "Об утверждении Правил формирования перечня специализированных продуктов лечебного питания для детей-инвалидов"
Приложение Б. Алгоритмы ведения пациента

Приложение В. Информация для пациентов
Фенилкетонурия – это наследственное заболевание, характеризующееся нарушением метаболизма фенилаланина (незаменимая аминокислота, которая не синтезируется в организме, поступает с пищей с продуктами животного происхождения, в том числе с грудным молоком и детскими молочными смесями).
При отсутствии своевременной диагностики и лечения заболевание проявляется обычно в возрасте 2-6 месяцев признаками поражения центральной нервной системы - родителей беспокоит вялость ребенка, отсутствие интереса к окружающему, иногда повышенная раздражительность, беспокойство, срыгивания, нарушение мышечного тонуса (чаще мышечная гипотония), признаки атопического дерматита, задержка психомоторного развития, иногда судороги. С возрастом дети имеют тяжелое поражение нервной системы, вплоть до умственной отсталости и эпилепсии. При своевременно назначенном патогенетическом лечении жалобы имеют более легкий характер или отсутствуют.
С целью ранней диагностики и профилактики развития указанных симптомов проводится неонатальный скрининг, результаты которого сообщаются родителям по телефону.
Родители обязаны внимательно отнестись к полученной из центра неонатального скрининга информации и немедленно явиться на прием к врачу и/или связаться с ним по телефону, даже если у ребенка отсутствуют клинические симптомы. Решение о назначении и характере диетотерапии принимает врач.
Диетотерапия, основанная на резком ограничении фенилаланина в рационе больных детей за счет исключения высокобелковых продуктов, должна быть начата не позднее первых недель жизни ребенка с целью достижения максимальной эффективности лечения. Недостающее количество белка восполняется за счет специализированных лечебных продуктов, частично или полностью лишенных фенилаланина.
Назначение патогенетической диетотерапии с первых дней жизни ребенка определяет благоприятный прогноз течения фенилкетонурии.
Учитывая аутосомно-рецессивный тип наследования и высокий риск повторного рождения в семье больного ребенка, показано медико-генетическое консультирование семей.
Приложение Г.
Приложение Г1. Схема метаболических процессов, приводящих к развитию гиперфенилаланинемии (фенилкетонурии)
.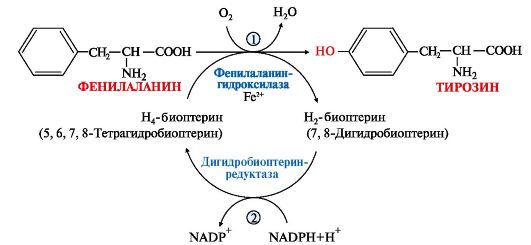
Рис.1 - Реакция 1- преобразование фенилаланина в тирозин под действием фермента фенилаланингидроксилазы (ФАГ); 2-Тетрагидробиоптерин (в присутствии Fe2+ под действием фермента дигидробиоптеринредуктазы окисляется до образования дигидробиоптерна).
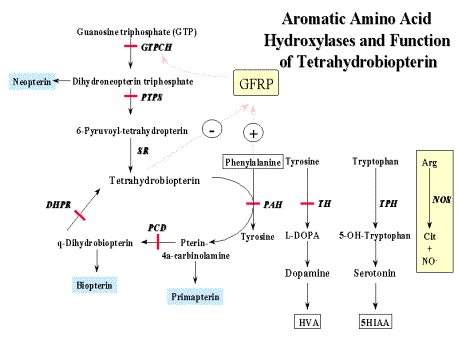
Роль тетрагидробиоптерина в гидроксилировании ароматических аминокислот
Рис.2 - Метаболизм тетрагидробиоптерина.
Приложение Г2. Схема неонатального скрининга на фенилкетонурию [11].

Приложение Г3. Правила взятия крови у новорожденного
Взятие крови у новорожденного проводится на 4-5-й день жизни (у недоношенных детей на 7-й день жизни), но не ранее чем через 2 дня после начала энтерального питания. Взятие крови ранее 4-х дней жизни нежелателен из-за большого числа ложноположительных и ложноотрицательных результатов тестирования.
Образец крови берут из пятки новорожденного ребенка через 3 часа после кормления. Предварительно необходимо согреть стопу ребёнка, обернув её в полотенце, смоченное теплой водой (не выше 42 градусов) на 1-2 минуты, затем протереть область пункции стерильной салфеткой, смоченной 70% спиртом. Во избежание гемолиза крови обработанное место следует промокнуть сухой стерильной салфеткой.
Место прокола расположено медиально от линии, проведенной от большого пальца до пятки, или латерально от линии, проведенной от мизинца до пятки (заштрихованная область на рис. 1). Глубина пунктирования не должна превышать 2-5 мм, т.к. при более глубоком проколе возникает опасность остеомиелита.

Рис. 1. Взятие образца крови из пятки новорожденного
Прокол осуществляется одноразовым скарификатором, первая капля крови снимается стерильным сухим тампоном. Вторая капля крови наносится на тест-бланк, пропитываемый кровью полностью и насквозь в указанных местах (кружки). Чрезмерное сдавливание места прокола может вызвать гемолиз или примешивание к образцу тканевой жидкости.
Кровь наносится только на лицевую сторону бланка. На каждую область кровь наносится только один раз. Запрещено наслаивать на уже нанесенную кровь второй слой, это искажает результаты исследования. Если крови мало, лучше правильно заполнить меньше областей, чем все, но неправильно.
В связи с тем, что скрининг проводится на несколько болезней обмена, пятен должно быть не менее пяти. Вид пятен должен быть одинаковым с обеих сторон тест-бланка.
Бланк с кровью высушивается в горизонтальном положении на чистой обезжиренной поверхности в течение 3 часов при комнатной температуре (15-220С). Не допускается соприкосновение бланков между собой во время сушки. Нельзя использовать любые виды нагрева бланка для ускорения сушки (солнечный свет, фен, батарея и т.п.).
В обменной карте новорожденного делается отметка о проведенном заборе крови. В случае отсутствия в документации новорожденного ребенка отметки о заборе образца крови, при его поступлении под наблюдение в детскую поликлинику по месту жительства или переводе по медицинским показаниям в больничное учреждение забор образца крови осуществляется в указанных медицинских учреждениях в соответствии с рекомендациями. Высушенный тест-бланк с образцом крови отправляется с соблюдением температурного режима в лабораторию неонатального скрининга.
Неправильная подготовка пациента к взятию крови, нарушение техники взятия пробы, правил хранения и транспортировки биологического материала, может привести к ошибочным результатам лабораторных исследований. Ложноположительные результаты скрининга приводят к увеличению количества необходимых повторных исследований и расхода реактивов, к неоправданному психологическому стрессу у родителей. Ложноотрицательные результаты могут привести к пропуску скринируемого заболевания. В процессе наблюдения за ребенком педиатр может выявить клинические симптомы, характерные для наследственных болезней обмена, даже при отрицательных результатах скрининга. В этом случае следует провести углубленное обследование ребенка.
Приложение Г4. Химический состав специализированных продуктов лечебного питания для больных ФКУ (в 100 г сухого продукта, для жидких продуктов указано отдельно)
|
Наименование продукта |
Белок (эквивалент), г |
Жир, г |
Углеводы, г |
Энергетическая ценность, ккал |
|
Продукты для детей первого года жизни |
||||
|
Афенилак 131 |
13 |
25 |
52 |
485 |
|
Афенилак 151 |
15 |
23 |
51,7 |
474 |
|
Нутриген 14 – phe PREMIUM1 |
14 |
23 в т.ч. ДЦПНЖК |
52 |
460 |
|
Анамикс ХР LCP |
|
|
|
|
|
МD мил ФКУ-0 |
13 |
23 в т.ч. ДЦПНЖК |
59 |
495 |
|
COMIDA-PKU А формула + LCP |
11,8 |
27,4 в т.ч. ДЦПНЖК |
52,6 |
506 |
|
Продукты для больных ФКУ старше 1 года |
||||
|
Афенилак 201 |
20 |
18 |
50,4 |
444 |
|
Афенилак 401 |
40 |
13,5 |
31 |
405 |
|
Нутриген 301 |
30 |
0 |
54 |
336 |
|
Нутриген 701,2 |
70 |
0 |
6,9 |
308 |
|
Нутриген 751,2 |
75 |
0 |
1,3 |
305 |
|
Нутриген 20 – phe PREMIUM1 |
20 |
18 |
48 |
434 |
|
Нутриген 40 – phe PREMIUM1 |
40 |
13,5 |
28 |
393 |
|
Нутриген 70 – phe PREMIUM1,2 |
70 |
0 |
0-12 |
280 |
|
П-АМ 1 |
75 |
0 |
0 |
300 |
|
П-АМ 2 |
75 |
0 |
0 |
300 |
|
П-АМ 32 |
75 |
0 |
0 |
300 |
|
ХР Максамейд |
39 |
менее 0,5 |
34 |
297 |
|
ХР Максамум |
25 |
менее 0,5 |
51 |
309 |
|
Изифен (100 мл) |
6,7 |
2,0 |
5,1 |
65 |
|
Лофлекс (62,5мл) |
10 |
- |
4,4 |
58 |
|
П-АМ материнский2 |
77,5 |
0 |
0 |
310 |
|
PKU Nutri 2 Energy |
27 |
14 |
42 |
402 |
|
МD мил ФКУ-1 |
20 |
0 |
73 |
372 |
|
МD мил ФКУ-2 |
40 |
6,1 |
46,9 |
402 |
|
МD мил ФКУ-32 |
69,1 |
0 |
23 |
368 |
|
МD мил ФКУ Премиум2 |
69,1 |
1,9 |
23 |
385 |
|
COMIDA-PKU В формула |
31,1 |
15 |
40,6 |
422 |
|
COMIDA-PKU В формула Клубника |
31,1 |
14,2 |
40,5 |
419 |
|
COMIDA-PKU В формула Карамель |
31,1 |
14,8 |
40,6 |
422 |
|
COMIDA-PKU В формула Шоколад |
31,1 |
14,2 |
38,7 |
415 |
|
COMIDA-PKU В формула Апельсин-Лимон |
31,1 |
13,1 |
41,1 |
412 |
|
COMIDA-PKU В2 |
73 |
0 |
0,5 |
296 |
|
COMIDA-PKU С формула |
45 |
0 |
38,9 |
335 |
|
COMIDA-PKU С формула Апельсин-Лимон |
45 |
0 |
36 |
331 |
|
COMIDA-PKU С2 |
75 |
0 |
0,4 |
302 |
|
COMIDA-PKU С2 капсулы |
75 |
0 |
0,4 |
302 |
-
специализированные продукты отечественного производства .
-
специализированные продукты, которые могут использоваться в диетотерапии женщин с ФКУ в период преконцепционной подготовки и во время беременности
Приложение Г5. Тест на чувствительность к сапроптерину дигидрохлориду
Тестирование потенциальной чувствительности к сапроптерину и дальнейшее лечение им проводит и контролирует врач, который осуществляет наблюдение пациентов с ФКУ. Для более тщательного контроля тест целесообразно проводить в стационаре, возможно дневного пребывания, при удовлетворительной комплаентности пациента – амбулаторно [4,5,16,20].
Цель тестирования – выявить ВН4 зависимые формы ГФА и определить чувствительность к сапроптерином у больных с классической ФКУ.
Тестирование возможно проводить по 2-х дневной или 7-ми дневной схеме [25,26].
Дозировка препарата во время тестирования составляет от 5 до 20 мг/кг массы тела (в среднем 10 мг/кг массы тела), некототорые авторы рекомендуют максимальную дозировку для проведения тестирования [26]. Суточная доза рассчитывается индивидуально для каждого пациента в соответствии с инструкцией к препарату.
Перед проведением тестирования стабилизируют содержание фенилаланина в крови путем поддержания постоянства пищевого рациона в течение 4-5 дней до начала теста и в период тестирования.
Тестирование начинают на фоне стабильного уровня ФА крови, при этом рассчитывается среднее значение двух показателей ФА, полученных в результате двукратного взятия крови перед началом тестирования, среднее значение должно быть не ниже 450 мкмоль/л (7,5 мг%).
В день первого приема препарата натощак берут анализ крови из пальца на фенилаланин, затем ребенок принимает рассчитанную дозу препарата вместе с завтраком или сразу же после завтрака. В течение 2-х (или 7-ми) дней утром в одно и то же время после завтрака (или вместе с едой) ребенок принимает препарат, разведенный в теплой воде в соответствии с инструкцией.
Повторное взятие крови проводят на 3-й (или 8-й) день строго натощак. Ответ на чувствительность к препарату оценивается по степени снижения концентрации фенилаланина в крови больного при соблюдении стабильной гипофенилаланиновой диеты. Пациент считается чувствительным, если разница уровня ФА, полученного по окончании периода оценки ответа на лечение, и исходного уровня ФА перед началом приема препарата составляет 30% и более.
При снижении уровня ФА более 85% высока вероятность, что у пациента ГФА, обусловленная недостаточностью тетрагидробиоптерина (ВН4).
Важным условием тестирования является стабильная гипофенилаланиновая диета с целью исключения ее влияния на колебания концентрации ФА в крови.
Схемы тестирования приведены в таблицах 1-2.
Таблица 1 - Двухдневная схема тестирования для определения чувствительности к сапроптерину.

Таблица 2 - Семидневная схема тестирования для определения чувствительности к сапроптерину

Повышение толерантности к пищевому ФА на фоне приема сапроптерина является показанием для расширения диеты, при этом концентрация ФА в крови не должна превышать 6 мг% (360 мкмоль/л). При комбинированном использовании гипфенилаланиновой диеты и сапроптерина контроль за антропометрическими и биохимическими показателями нутритивного статуса должны быть систематическим во избежание развития дефицита основных и минорных пищевых веществ, алиментарно-зависимых состояний.
При необходимости длительная медикаментозная терапия у больных ФКУ, отвечающих на лечение сапроптерина дигидрохлоридом снижением уровня ФА в крови, проводится в комбинации с диетой при использовании аминокислотных смесей, количество которых определяет врач.
После проведения тестирования и получения положительного результата необходимо подтвердить его в ходе длительного наблюдения. Пациенты, получающие сапроптерин требуют тщательного контроля за состоянием их нутритивного статуса и другими показателями здоровья.
Медикаментозная терапия при ВН4-дефицитных состояниях с ГФА и без ГФА: начальная доза сапроптерина дигидрохлорида у больных с недостаточностью BH4 составляет от 2 до 5 мг/кг массы тела при приеме 1 раз в день. При необходимости доза сапроптерином может быть увеличена до 20 мг/кг массы тела в день, для достижения оптимального терапевтического эффекта возможен прием препарата 2 -3 в день.
Приложение Г6. Расшифровка примечаний
…ж – лекарственный препарат, входящий в Перечень жизненно необходимых и важнейших лекарственных препаратов для медицинского применения на 2016 год (Распоряжение Правительства РФ от 26.12.2015 N 2724-р).
Комментарии
ПРАКТИКА ПЕДИАТРА



