Варгатеф - инструкция по применению
Синонимы, аналоги СтатьиРегистрационный номер:
ЛП-002830
Торговое наименование:
ВАРГАТЕФ
Международное непатентованное наименование:
нинтеданиб (nintedanib)
Лекарственная форма:
мягкие капсулы
Состав
Одна капсула 100 мг содержит:
Активное вещество: нинтеданиба этансульфонат - 120,40 мг, что соответствует 100,00 мг нинтеданиба основания;
Вспомогательные вещества: триглицериды средней цепи - 107,40 мг, твердый жир - 51,00 мг, лецитин - 1,20 мг;
Оболочка капсулы: желатин - 111,24 мг, глицерин 85% - 49,09 мг, титана диоксид (Е171) - 1,21 мг, железа оксид красный (суспензия с глицерином 85%) - 0,06 мг, железа оксид желтый (суспензия с глицерином 85 %) - 0,36 мг, чернила Опакод® тип S-1 -17823.
Одна капсула 150 мг содержит:
Активное вещество: нинтеданиба этансульфонат - 180,60 мг, что соответствует 150,00 мг нинтеданиба основания;
Вспомогательные вещества: триглицериды средней цепи - 161,10 мг, твердый жир -76,50 мг, лецитин - 1,80 мг;
Оболочка капсулы: желатин - 141,84 мг, глицерин 85% - 61,43 мг, титана диоксид (Е171) - 0.39 мг, железа оксид красный (суспензия с глицерином 85%) - 1,24 мг, железа оксид желтый (суспензия с глицерином 85 %) - 1,86 мг, чернила Опакод® тип S-1-17823.
Описание
Капсулы 100 мг
Продолговатые непрозрачные мягкие желатиновые капсулы розовато-оранжевого (персикового) цвета, содержащие ярко-желтую вязкую суспензию, на одной стороне которых черным цветом напечатан символ компании Берингер Ингельхайм и «100».
Капсулы 150 мг
Продолговатые непрозрачные мягкие желатиновые капсулы коричневого цвета, содержащие ярко-желтую вязкую суспензию, на одной стороне которых черным цветом напечатан символ компании Берингер Ингельхайм и «150».
Фармакотерапевтическая группа
противоопухолевое средство - протеинтирозинкиназы ингибитор
Код ATX:
L01XE31
Фармакологические свойства
Фармакодинамика
Механизм действия
Нинтеданиб - ингибитор тирозинкиназы, относящийся к классу малых молекул, блокирующий киназную активность рецепторов фактора роста эндотелия сосудов 1-3 (VEGFR 1-3), рецепторов тромбоцитарного фактора роста а и В (PDGFR а и В) и рецепторов фактора роста фибробластов 1-3 (FGFR 1-3). Кроме того, ингибируются Fms-подобная протеинтирозинкиназа (Flt-З), лимфоцит-специфическая протеинтирозинкиназа (Lck), протоонкогенная протеинтирозинкиназа (Src) и киназы рецептора колониестимулирующего фактора 1 (CSF1R). Нинтеданиб конкурентно взаимодействует с аденозинтрифосфат (АТФ) связывающим участком этих киназ и блокирует каскады внутриклеточной передачи сигналов, которые, как было продемонстрировано, участвуют в патогенезе ремоделирования фиброзной ткани.
Фармакодинамические эффекты
Опухолевый ангиогенез является процессом, который вносит существенный вклад в рост, прогрессирование и образование метастазов опухоли. Этот процесс преимущественно запускается проангиогенными факторами такими, как фактором роста эндотелия сосудов и основным фактором роста фибробластов (VEGF и bFGF), секретируемыми опухолевыми клетками, чтобы привлечь эндотелиальные и периваскулярные клетки хозяина и облегчить доставку кислорода и питательных веществ через сосудистую систему.
Нинтеданиб эффективно противодействует образованию и развитию сосудистой системы опухоли, приводит к замедлению и остановке роста опухоли.
В исследованиях in vitro на клетках человека было показано, что нинтеданиб ингибирует процессы, предположительно связанные с инициацией фиброзного патогенеза, с высвобождением профибротических медиаторов из моноцитов периферической крови и с поляризацией макрофагов с образованием макрофагов. активированных по альтернативному пути. Было продемонстрировано, что нинтеданиб ингибирует фундаментальные процессы фиброза органов, пролиферации и миграции фибробластов, трансформацию в активный фенотип миофибробласта и секрецию внеклеточного матрикса. В исследованиях на животных с использованием многочисленных моделей ИЛФ (идиопатический легочный фиброз), ИЗЛ-ССД (интерстициальные заболевания легких при системной склеродермии), ИЗЛ-РА (интерстициальные заболевания легких при ревматоидном артрите) и других видов фиброза органов, было показано противовоспалительное и противофибротическое действие нинтеданиба в легких, коже, сердце, почках и печени. Нинтеданиб также обладает активностью в отношении сосудов. Он уменьшал апоптоз эндотелиальных клеток микрососудов кожи и замедлял ремоделирование легочных сосудов путем уменьшения пролиферации клеток гладкой мускулатуры сосудов, толщины стенки легочных сосудов и уменьшал процентную долю окклюзированных легочных сосудов.
Фармакокинетика
Фармакокинетика нинтеданиба может считаться линейной в отношении времени (т.е. данные, полученные в случае применения однократной дозы, могут экстраполироваться на данные, полученные в результате многократного применения). После многократного применения препарата накопление составляет 1,04 при оценке максимальной концентрации (Cmax) и 1,38 при оценке площади под кривой «концентрация-время» (AUCτ ). Остаточные концентрации нинтеданиба остаются стабильными в течение одного года и более.
Всасывание
Сmах нинтеданиба в плазме крови достигается приблизительно через 2-4 часа после приема препарата во время еды (диапазон 0,5-8 часов). Абсолютная биодоступность дозы 100 мг составляет у здоровых добровольцев 4,69 % (90 % доверительный интервал (ДИ): 3,615-6,078).
Установлено, что экспозиция нинтеданиба увеличивается пропорционально дозе (в диапазонах доз 50-450 мг один раз в день и 150-300 мг два раза в день). Устойчивые концентрации в плазме крови достигаются, как максимум, в течение одной недели после начала приема.
Экспозиция нинтеданиба увеличивается после приема пищи примерно на 20 % по сравнению с приемом препарата натощак (ДИ: 95,3-152,5 %), а всасывание замедляется. Медиана времени достижения максимальной концентрации в плазме крови натощак - 2,00 часа, после еды - 3,98 часа.
Распределение
Распределение нинтеданиба осуществляется путем двухфазной кинетики. После внутривенной инфузии во время терминальной фазы наблюдается большой объем распределения (Vd) 1050 л, геометрический коэффициент вариации (gCV) 45,0 %.
Связывание нинтеданиба с белками плазмы человека in vitro считается значительным, связанная фракция составляет 97,8 %. Основным белком, участвующим в связывании, считается альбумин сыворотки крови. Нинтеданиб преимущественно распределяется в плазме, соотношение кровь/плазма составляет 0,869.
Нинтеданиб является субстратом для гликопротеина-Р (P-gp). Показано, что нинтеданиб in vitro не является субстратом или ингибитором полипептидных переносчиков органических анионов (ОАТР-1В1, ОАТР-1ВЗ, ОАТР-2В1), переносчика органических катионов-2 (ОСТ-2) или белка, ассоциированного с множественной лекарственной устойчивостью-2 (MRP-2). Нинтеданиб также не является субстратом для белка резистентности рака молочной железы (BCRP). In vitro было установлено, что нинтеданиб обладает слабой ингибирующей активностью в отношении ОСТ-1, BCRP и P-gp, что, как считается, имеет небольшую клиническую значимость. Такое же заключение сделано в отношении нинтеданиба как субстрата для ОСТ-1.
Биотрансформация
Основной реакцией, участвующей в метаболизме нинтеданиба, является гидролитическое расщепление с помощью эстераз, что приводит к образованию свободного кислого метаболита нинтеданиба. В доклинических исследованиях in vivo установлено, что данный метаболит не обладает эффективностью, несмотря на влияние на целевые рецепторы препарата. В дальнейшем он глюкуронизируется ферментами уридин-дифосфат-глюкуронозилтрансферазами (UGT 1А1, UGT 1А7, UGT 1А8 и UGT 1А10) с образованием глюкуронида.
Считается, что биотрансформация нинтеданиба с участием изоферментов СУР происходит только в небольшой степени, основное участие принимает изофермент CYP ЗА4. В исследовании ADME у человека основной метаболит, образующийся с участием изоферментов CYP, обнаружить в плазме не удалось. По данным исследования in vitro, CYP-зависимый метаболизм составляет примерно 5 %, тогда как расщепление, осуществляемое эстеразами, составляет 25 %.
Выведение
Общий клиренс из плазмы после внутривенной инфузии составляет 1390 мл/мин (gCV 28,8 %). Экскреция почками неизмененной активной субстанции в течение 48 часов после приема нинтеданиба внутрь составляет примерно 0,05 % от величины дозы (gCV 31,5 %), а после внутривенного введения - примерно 1,4 % (gCV 24,2 %); почечный клиренс составляет 20 мл/мин (gCV 32,6%). После приема внутрь [ 14С]-нинтеданиба радиоактивный материал выводился преимущественно с желчью и обнаруживался в кале (93,4% от величины дозы, gCV 2,61 %). Вклад почечной экскреции в общий клиренс составляет 0.649 % от величины дозы (gCV 26,3 %). Выведение считается полным (более 90 %) через 4 дня после приема. Период полувыведения нинтеданиба составляет от 10 до 15 часов (gCV примерно 50 %).
Зависимость эффекта от экспозиции
Немелкоклеточный рак легкого (НМРЛ)
В ходе исследования зависимости нежелательных явлений от фармакокинетики нинтеданиба была установлена тенденция к более частому повышению активности ферментов печени при большей экспозиции нинтеданиба. Подобной зависимости не отмечается для нарушений со стороны желудочно-кишечного тракта (ЖКТ).
Для клинических конечных точек анализ взаимосвязи фармакокинетики с эффективностью не проводился. С помощью метода логистической регрессии установлена статистически значимая взаимосвязь между экспозицией нинтеданиба и влиянием на изменения, обнаруживаемые методом динамической контрастной магнитно-резонансной томографии.
ИЛФ и ИЗЛ-ССД
Анализ зависимости «экспозиция-ответ» показал наличие взаимосвязи типа Еmах между воздействием в диапазоне, наблюдавшемся в ряде исследований Фазы II и III, и ежегодным уровнем снижения форсированной жизненной емкости легких; ЕС50 составляла около 3-5 нг/мл (относительная стандартная ошибка: приблизительно 55 %). Для сравнения, медиана наблюдаемых минимальных концентраций нинтеданиба при применении препарата ВАРГАТЕФ в дозе 150 мг 2 раза в сутки составляла примерно 10 нг/мл.
В отношении безопасности возможно имеется слабая взаимосвязь между экспозицией нинтеданиба в плазме крови и повышением активности аланинаминотранферазы (АЛТ) и/или аспартатаминотрансферазы (ACT). Фактически применяемая доза может быть более существенным прогностическим фактором риска развития диареи любой интенсивности, даже если экспозицию в плазме крови нельзя исключить в качестве определяющего фактора риска.
Фармакокинетика у особых групп пациентов
Фармакокинетические свойства нинтеданиба были сопоставимы у здоровых добровольцев, пациентов с ИЛФ, ИЗЛ-ССД и НМРЛ. Основываясь на результатах популяционного фармакокинетического анализа и описательных исследований, на воздействие нинтеданиба не влиял пол (с учетом массы тела), нарушения функции почек легкой и средней степени тяжести (рассчитанная по клиренсу креатинина), метастазы в печени, общее состояние больного (оценка по опроснику Восточной Объединенной Онкологической Группы, Eastern Cooperative Oncology Group (ECOG)), употребление алкоголя, или генотип P-gp. Популяционный фармакокинетический анализ выявил умеренное влияние возраста, массы тела и расовой принадлежности на воздействие нинтеданиба. В связи с тем, что наблюдалась высокая межиндивидуальная вариабельность воздействия, эти влияния не считались клинически значимыми. Тем не менее, рекомендуется тщательное мониторирование пациентов, у которых выявляется несколько таких факторов риска, представленных ниже.
Возраст
Экспозиция нинтеданиба линейно увеличивается с возрастом. У 45-летних пациентов (5-й процентиль) значение AUCτ,ss (в равновесном состоянии на период дозирования) было ниже на 16 %, а у 76-летних пациентов (95-й процентиль) выше на 13 % по сравнению с пациентами, медиана возраста которых составляла 62 года. Диапазон возраста, оценивавшийся в ходе анализа, составлял 29-85 лет; возраст более 75 лет отмечался примерно у 5 % популяции пациентов.
Аналогичные исследования у детей не проводились.
Масса тела
Наблюдается обратная корреляция между массой тела и экспозицией нинтеданиба. У пациентов с массой тела 50 кг (5-й процентиль) величина AUCτ,ss увеличивалась на 25 %, а у пациентов с массой тела 100 кг (95-й процентиль) уменьшалась на 19 % по сравнению с пациентами, медиана массы тела которых составляла 71,5 кг.
Раса
Средняя популяционная экспозиция нинтеданиба на 33-50 % выше у пациентов-китайцев, тайваньцев и индийцев, и на 16% выше у пациентов японского происхождения, а у пациентов-корейцев - на 16-22 % ниже, чем у пациентов европеоидной расы (с поправкой на массу тела). Данные в отношении пациентов негроидной расы являются очень ограниченными, диапазон этих данных сходен с пациентами европеоидной расы.
Нарушения функции печени
В исследовании I фазы при изучении однократного введения препарата значения Сmах и AUC были в 2,2 раза выше у добровольцев с легкими нарушениями функции печени (класс А по шкале Чайлд-Пью; 90 % доверительный интервал (ДИ) 1,3-3,7 для Сmax и 1,2-3,8 для AUC, соответственно), чем у здоровых добровольцев.
У добровольцев с умеренными нарушениями функции печени (класс В по шкале Чайлд-Пью) значение Сmах (90 % ДИ 4,4-13,2) было в 7,6 раза выше, и 8,7 раза выше значение AUC (90% ДИ 5,7-13,1) соответственно, в сравнении со здоровыми добровольцами. В исследовании не принимали участия пациенты с тяжелыми нарушениями функции печени (класс С по шкале Чайлд-Пью).
Показания к применению
- Местнораспространенный, метастатический или рецидивирующий немелкоклеточный рак легкого (аденокарцинома) после химиотерапии первой линии в комбинации с доцетакселом.
- Идиопатический легочный фиброз (идиопатический фиброзирующий альвеолит); для лечения и замедления прогрессирования заболевания.
- Интерстициальные заболевания легких при системной склеродермии
Противопоказания
- Гиперчувствительность к нинтеданибу, сое или арахису, или любому вспомогательному компоненту препарата;
- беременность и период грудного вскармливания;
- нарушения функции печени средней и тяжелой степени тяжести (опыт применения отсутствует);
- тяжелые нарушения функции почек (клиренс креатинина < 30 мл/мин) (опыт применения отсутствует)*;
- активные метастазы в головной мозг (опыт применения отсутствует);
- возраст младше 18 лет (опыт применения отсутствует).
* Эффективность и безопасность нинтеданиба у пациентов с тяжелыми нарушениями функции почек (клиренс креатинина < 30 мл/мин) не были изучены. Через почки выводится менее 1 % разовой дозы нинтеданиба.
В отношении противопоказаний для доцетаксела, пожалуйста, обратитесь к соответствующей инструкции по применению этого препарата.
С осторожностью
Нарушения функции печени легкой степени тяжести; наследственная предрасположенность к кровотечениям (болезнь фон Виллебранда); стабильные метастазы в головной мозг; терапия антикоагулянтами; венозные тромбоэмболии; перфорации ЖКТ в анамнезе; пациенты, которые ранее подвергались абдоминальным хирургическим вмешательствам; артериальная тромбоэмболия.
Применение при беременности и в период грудного вскармливания
Беременность
Специальных исследований по применению препарата ВАРГАТЕФ во время беременности у человека не проводилось, однако в доклинических исследованиях у животных установлена репродуктивная токсичность этого препарата. Поскольку нинтеданиб может обладать эмбриотоксическим действием у человека, он не должен применяться во время беременности, поэтому перед началом лечения необходимо проведение теста на беременность, а также во время терапии по мере необходимости. Пациенткам следует немедленно сообщить врачу о развитии беременности во время терапии препаратом ВАРГАТЕФ. Если во время терапии развивается беременность, необходимо прекратить лечение препаратом ВАРГАТЕФ и проинформировать пациентку о потенциальной опасности эмбриотоксического воздействия препарата. Применение нинтеданиба может быть вредно для плода. Женщинам детородного возраста, принимающим препарат ВАРГАТЕФ, следует рекомендовать избегать наступления беременности во время лечения препаратом ВАРГАТЕФ и использовать высокоэффективные методы контрацепции во время применения препарата и на протяжении, по крайней мере, 3 месяцев после приема последней дозы. В данный момент неизвестно, может ли нинтеданиб снижать эффективность гормональных контрацептивов, поэтому женщинам, использующим гормональные контрацептивы, необходимо использовать и барьерные методы контрацепции.
Грудное вскармливание
Специальных исследований у человека о выделении нинтеданиба и его метаболитов в грудное молоко не проводилось. В доклинических исследованиях показано, что у крыс в период лактации в грудное молоко проникает небольшое количество нинтеданиба и его метаболитов (≤ 0,5 % от величины применявшейся дозы). Поэтому нельзя исключить риск для новорожденных и грудных детей. Во время лечения препаратом ВАРГАТЕФ кормление грудью следует прекратить.
Фертильность
В доклинических исследованиях признаков нарушений фертильности у самцов выявлено не было. В субхронических и хронических исследованиях токсичности, во время которых уровень системного воздействия препарата был сравним с уровнем, достигающимся при использовании максимальной рекомендуемой дозы у человека, признаков нарушений фертильности у самок крыс обнаружено не было.
За информацией о влиянии доцетаксела на беременность, грудное вскармливание и фертильность, пожалуйста, обратитесь к соответствующей инструкции по применению этого препарата.
Способ применения и дозы
Капсулы принимают внутрь предпочтительно во время еды. Капсулы следует проглатывать целиком, запивая водой, не разжевывая и не разламывая.
Пропуск дозы
Если какая-либо доза препарата ВАРГАТЕФ была пропущена, то следует продолжить прием препарата в изначально рекомендуемой дозе по расписанию следующего приема препарата. Если доза была пропущена, пациент не должен принимать дополнительную дозу препарата.
НМРЛ
Лечение препаратом ВАРГАТЕФ следует назначать и проводить под контролем врача, имеющего опыт в назначении противоопухолевой терапии.
Рекомендуемая доза препарата ВАРГАТЕФ составляет 200 мг два раза в день с интервалом примерно в 12 часов со 2 по 21 день стандартного 21-дневного цикла лечения доцетакселом.
ВАРГАТЕФ не должен применяться в день начала химиотерапии доцетакселом, т.е. в 1 день лечения.
Максимальная рекомендуемая суточная доза составляет 400 мг.
После окончания применения доцетаксела можно продолжить терапию препаратом ВАРГАТЕФ до тех пор, пока сохраняется клинический эффект, или до развития неприемлемой токсичности.
Изменение дозы в случае развития нежелательных реакций
В качестве первоначальной меры для устранения нежелательных реакций рекомендуется временный перерыв в лечении препаратом ВАРГАТЕФ до тех пор, пока нежелательная реакция не снизится до уровня, который позволит возобновить терапию (см. Таблицы 1 и 2). Лечение может возобновляться в уменьшенной дозе.
Для обеспечения индивидуальной безопасности и переносимости рекомендуется снижение суточной дозы препарата на 100 мг (т.е. уменьшение разовой дозы на 50 мг), как это описывается в Таблицах 1 и 2. Если нежелательная реакция (реакции) сохраняется, т.е. если пациент не переносит препарат в дозе 100 мг два раза в день, лечение препаратом нужно окончательно прекратить.
В случае специфического повышения активности АСТ/АЛТ более чем в 3 раза по сравнению с ВГН в сочетании с повышением концентрации общего билирубина в 2 и более раза по сравнению с ВГН и щелочной фосфатазы (ЩФ) менее чем в 2 раза по сравнению с ВГН (см. Таблицу 2), применение препарата ВАРГАТЕФ следует временно прекратить. Если не будет установлена альтернативная причина нарушений, ВАРГАТЕФ должен быть окончательно отменен.
Таблица 1.
Информация об изменении дозы в случае развития диареи, рвоты и других негематологических или гематологических нежелательных явлений, исключая повышение уровней печеночных ферментов (см. Таблицу 2).
Нежелательная реакция* | Изменение дозы |
Диарея 2 степени тяжести в течение более 7 дней подряд, несмотря на антидиарейное лечение. | После временного прекращения лечения и уменьшения тяжести реакции до 1 степени или восстановления исходного состояния пациента рекомендуется снижение дозы с 200 мг два раза в день до 150 мг два раза в день. |
Рвота ≥ 2 степени тяжести |
* Общие терминологические критерии для нежелательных явлений - СТСАЕ (Common Terminology Criteria for Adverse Events)
Таблица 2.
Информация об изменении дозы в случае повышения активности ACT и/или АЛТ и повышения концентрации билирубина.
Повышение АСТ/АЛТ и билирубина | Изменение дозы |
Повышение активности ACT и/или АЛТ в > 2,5 раза по сравнению с ВГН в сочетании с повышением общего билирубина в ≥ 1,5 раза по сравнению с ВГН. | После временного прекращения лечения и снижения активности трансаминаз до ≤ 2,5 раза по сравнению с ВГН в сочетании со снижением билирубина до нормы, доза снижается с 200 мг 2 раза в день до 150 мг 2 раза в день и, если еще одно снижение дозы считается необходимым, то со 150 мг 2 раза в день до 100 мг два раза в день. |
Повышение значений ACT и/или АЛТ в > 3 раза по сравнению с ВГН в сочетании с повышением общего билирубина в ≥ 2 раза по сравнению с ВГН и повышением ЩФ в < 2 раза по сравнению с ВГН. | Если альтернативная причина нарушений не будет установлена, то препарат ВАРГАТЕФ должен быть окончательно отменен. |
ИЛФ и ИЗЛ-ССД
Лечение препаратом ВАРГАТЕФ следует назначать и проводить под контролем врача, имеющего опыт диагностики и лечения заболеваний, при которых показан препарат. Рекомендуемая доза препарата составляет 150 мг два раза в день, приблизительно через каждые 12 часов.
Максимальная суточная доза составляет 300 мг.
Изменение дозы в случае развития нежелательных реакций
При развитии нежелательных реакций таких, как диарея, тошнота и рвота в дополнение к симптоматической терапии, при необходимости, рекомендуется снижение дозы или временное прерывание лечения до тех пор, пока нежелательная реакция не снизится до уровня, который позволит возобновить терапию. Лечение препаратом ВАРГАТЕФ может быть возобновлено в полной дозе (150 мг два раза в день) или в сниженной дозе (100 мг два раза в день). Если пациент не переносит дозу препарата 100 мг два раза в день, лечение препаратом ВАРГАТЕФ следует прекратить.
В случае прерывания лечения вследствие повышения активности трансаминаз (ACT или АЛТ) более чем в 3 раза по сравнению с ВГН после восстановления показателей до нормальных значений рекомендовано возобновить терапию в уменьшенной дозе (100 мг два раза в день). Впоследствии доза может быть увеличена до полной дозы (150 мг два раза в день).
Особые группы пациентов
Детский возраст
Безопасность и эффективность препарата ВАРГАТЕФ у пациентов детского возраста в клинических исследованиях не изучалась.
Пожилой возраст (>65 лет)
Не отмечено никаких общих различий по безопасности и эффективности применения препарата у пожилых пациентов по сравнению с пациентами моложе 65 лет. Исходной коррекции дозы препарата на основании возраста пациента не требуется.
Раса и масса тела
Основываясь на данных популяционного фармакокинетического анализа, исходная коррекция дозы препарата ВАРГАТЕФ в зависимости от расы или массы тела не требуется. Данные по безопасности у пациентов негроидной расы ограничены.
Нарушения функции почек
Через почки выводится менее 1 % однократной дозы нинтеданиба. У пациентов с нарушениями функции почек легкой или средней степени тяжести изменения начальной дозы не требуется. У пациентов с тяжелыми нарушениями функции почек (клиренс креатинина < 30 мл/мин) безопасность, эффективность и фармакокинетика нинтеданиба не изучались.
Нарушения функции печени
Нинтеданиб выводится преимущественно с желчью (через кишечник) (> 90 %). У пациентов с нарушениями функции печени (класс А по шкале Чайлд-Пью, класс В по шкале Чайлд-Пью), концентрация препарата в крови повышалась.
У пациентов с нарушениями функции печени средней и тяжелой степени тяжести (классы В и С по шкале Чайлд-Пью) безопасность, эффективность и фармакокинетика нинтеданиба не изучались. Поэтому лечение пациентов с нарушениями функции печени средней и тяжелой степени тяжести препаратом ВАРГАТЕФ не рекомендуется.
НМРЛ
У пациентов с нарушениями функции печени легкой степени тяжести (класс А по шкале Чайлд-Пью), изменения начальной дозы не требуется.
ИЛФ и ИЗЛ-ССД
Для пациентов с нарушениями функции печени легкой степени тяжести (класс А по шкале Чайлд-Пью) рекомендованная доза препарата ВАРГАТЕФ составляет 100 мг два раза в день с интервалом примерно в 12 часов.
В случае пациентов с нарушениями функции печени легкой степени тяжести (класс А по шкале Чайлд-Пью) для устранения нежелательных реакций следует рассмотреть возможность временного перерыва в лечении препаратом или окончательного прекращения терапии.
За информацией о дозировании, способе применения и модификации дозы доцетаксела, пожалуйста, обратитесь к соответствующей инструкции по применению этого препарата.
Побочное действие
Частота побочных реакций, приведенных ниже, изложена в соответствии со следующей градацией: очень часто (≥ 1/10); часто (≥ 1/100; < 1/10); нечасто (≥ 1/1000; < 1/100); редко (≥ 1/10000; < 1/1000); очень редко (< 1/10000), неизвестно (частота не может быть оценена по доступным данным). Данные о побочных реакциях также учитывают опыт пострегистрационного применения.
НМРЛ
Наиболее часто сообщавшимися нежелательными реакциями, считавшимися связанными с применением препарата ВАРГАТЕФ, были диарея, повышение активности ферментов печени (АЛТ и ACT) и рвота.
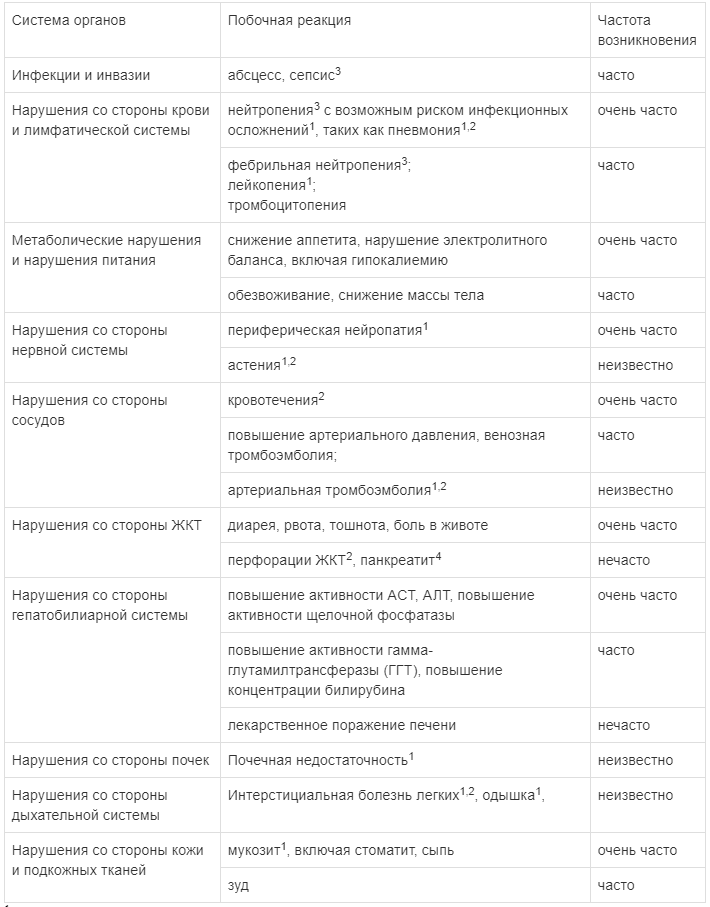 1 - Данные побочные реакции наблюдались при проведении клинических исследований, однако связь с приемом препарата ВАРГАТЕФ не доказана.
1 - Данные побочные реакции наблюдались при проведении клинических исследований, однако связь с приемом препарата ВАРГАТЕФ не доказана.
2 - В клинических исследованиях частота встречаемости данных побочных реакций у пациентов, принимавших нинтеданиб в комбинации с доцетакселом, не превышала частоту встречаемости таковых у пациентов, принимавших плацебо в комбинации с доцетакселом.
3 - Пожалуйста, обратитесь также к инструкции по медицинскому применению доцетаксела.
4 - Случаи панкреатита были зафиксированы у пациентов, принимавших нинтеданиб при лечении ИЛФ и НМРЛ. Большая часть этих случаев была зафиксирована у пациентов с показанием ИЛФ.
Диарея является часто сообщавшимся нежелательным явлением со стороны ЖКТ. Отмечается тесная временная связь между развитием диареи и применением доцетаксела. В клиническом исследовании LUME-Lung 1 диарея легкой и средней степени тяжести отмечалась у большинства пациентов. Диарея ≥ 3 степени тяжести при использовании комбинированного лечения отмечалась у 6,3 % пациентов, тогда как при использовании одного доцетаксела - только у 3,6 % пациентов.
Такие часто сообщающиеся побочные эффекты, как тошнота и рвота, в большинстве случаев были легкой или средней степени тяжести.
По данным клинических исследований, применение препарата ВАРГАТЕФ в комбинации с доцетакселом сопровождалось более частым развитием нейтропении ≥ 3 степени тяжести (по критериям СТСАЕ), чем в случае применения одного доцетаксела.
Повышение активности печеночных ферментов (АЛТ, ACT, ЩФ, гамма-глутамилтрансфераза (ГГТ)) и концентрации билирубина было в большинстве случаев обратимым при уменьшении дозы или прерывании терапии.
Ингибирование VEGFR способно привести к увеличению риска кровотечений. В клинических исследованиях у пациентов с аденокарциномой, принимавших ВАРГАТЕФ. повышение частоты кровотечений было сравнимо в обеих лечебных группах. Самым частым видом кровотечений было носовое кровотечение легкой или средней степени тяжести. Дисбаланса легочных или смертельных кровотечений не наблюдалось, о церебральных кровотечениях не сообщалось. Большинство летальных кровотечений были опухоль - ассоциированными.
В пострегистрационном периоде наблюдались случаи несерьезных и серьезных кровотечений, некоторые из которых приводили к летальному исходу. У пациентов с кровотечениями 3 или 4 степени тяжести следует тщательно оценить пользу и риски продолжения терапии препаратом ВАРГАТЕФ, а также необходимо рассмотреть возможность прекращения данной терапии. При возобновлении терапии препаратом ВАРГАТЕФ рекомендуется использовать сниженную суточную дозу.
Частота артериальных тромбоэмболий в двух лечебных группах исследования LUME-Lung 1 была сравнимой. Пациенты с недавно перенесенным инфарктом миокарда или инсультом из исследования исключались.
Частота перфораций ЖКТ в отдельных лечебных группах в клинических исследованиях была сопоставима.
ИЛФ и ИЗЛ-ССД
Наиболее часто сообщавшимися нежелательными реакциями, связанными с применением нинтеданиба, были диарея, тошнота и рвота, боль в области живота, снижение аппетита, снижение массы тела и повышение активности ферментов печени.
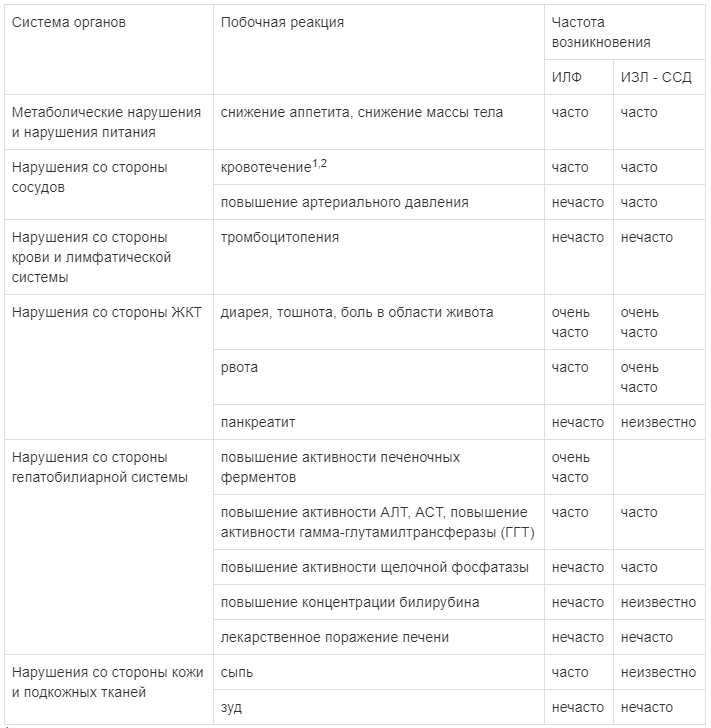 1 - Представленный термин описывает более широкую группу нежелательных явлений, нежели чем отдельное состояние или термин, предусмотренный MedDRA.
1 - Представленный термин описывает более широкую группу нежелательных явлений, нежели чем отдельное состояние или термин, предусмотренный MedDRA.
2 - В пострегистрационном периоде наблюдались случаи несерьезных и серьезных кровотечений, некоторые из которых приводили к летальному исходу.
В клинических исследованиях диарея была наиболее часто репортируемым желудочно-кишечным явлением. У большинства пациентов данные явления были легкой и средней степени тяжести и отмечались на протяжении первых 3 месяцев лечения. В исследованиях INPULSIS у пациентов с ИЛФ диарея отмечалась у 62,4 % по сравнению с 18,4% пациентов, получавших препарат ВАРГАТЕФ или плацебо, соответственно. Диарея стала причиной снижения дозы у 10,7 % пациентов, а прекращения терапии препаратом ВАРГАТЕФ - у 4,4 % пациентов. В исследовании SENSCIS у пациентов с ИЗЛ-ССД диарея отмечалась у 75,7 % в сравнении с 31,6 % пациентов, получавших препарат ВАРГАТЕФ и плацебо, соответственно. Диарея стала причиной снижения дозы препарата ВАРГАТЕФ у 22,2 % пациентов и причиной прекращения терапии препаратом у 6,9 % пациентов.
Тошнота и рвота были часто сообщаемыми нежелательными явлениями. У большинства пациентов отмечалась тошнота и рвота легкой или средней степени тяжести. Тошнота стала причиной прекращения лечения препаратом нинтеданиб у 2 % пациентов. Рвота стала причиной прекращения лечения у 0.8 % пациентов.
Ингибирование VEGFR может быть связано с повышением риска развития кровотечений. В клинических исследованиях частота развития кровотечений у пациентов была немного выше в группе препарата ВАРГАТЕФ (10,3% в исследовании INPULSIS, 11,1 % в исследовании SENSIS), по сравнению с группой плацебо (7.8 % в исследовании INPULSIS, 8,3 % в исследовании SENSIS). Наиболее частыми нежелательными явлениями в виде кровотечений были носовые кровотечения, не относившиеся к категории серьезных. Серьезные кровотечения возникали нечасто и с одинаковой частотой в обеих лечебных группах (плацебо -1,4%; ВАРГАТЕФ - 1,3%). В исследовании SENSCIS серьезные кровотечения возникали нечасто в обеих группах лечения (плацебо - 0,7 %, ВАРГАТЕФ - 1,4 %).
В пострегистрационном периоде наблюдались случаи несерьезных и серьезных кровотечений, некоторые из которых приводили к летальному исходу. Из клинических исследований исключались пациенты с инфарктом миокарда или инсультом в ближайшем анамнезе. В исследовании INPULSIS случаи развития артериальной тромбоэмболии встречались редко: у 0,7 % пациентов, получавших плацебо, и 2,5 % пациентов в группе, получавших препарат ВАРГАТЕФ. В то время как нежелательные реакции в виде ишемической болезни сердца были сравнимы в группах нинтеданиба и плацебо, пациентов, у которых развился инфаркт миокарда, оказалось больше в группе препарата ВАРГАТЕФ (1,6%) по сравнению с группой плацебо (0,5 %). В исследовании SENSCIS случаи развития артериальной тромбоэмболии встречались редко: у 0,7 % пациентов, получавших плацебо, и у 0,7 % пациентов в группе, получавшей препарат ВАРГАТЕФ. Инфаркт миокарда встречался редко в группе плацебо (0,7 %) и не встречался в группе пациентов, получавших препарат ВАРГАТЕФ.
Передозировка
Симптомы
В клинических исследованиях нинтеданиб изучался в наивысшей однократной дозе 450 мг один раз в день. Также зафиксированы случаи передозировки при применении препарата в максимальной дозе 600 мг в течение восьми дней. Наблюдавшиеся нежелательные явления были сопоставимы с известным профилем безопасности нинтеданиба: увеличение активности ферментов печени и нарушения со стороны ЖКТ. Пациенты полностью восстановились после нежелательных явлений. В исследованиях INPULSIS был зафиксирован один случай непреднамеренного повышения дозы до 600 мг 2 раза в день на протяжении 21 дня у пациента с ИЛФ. В период некорректного приема препарата было зафиксировано развитие несерьезного нежелательного явления (назофарингит), которое купировалось в данном периоде без регистрации каких-либо других нежелательных реакций.
Лечение
Специфического антидота на случай передозировки нет. При подозрении на передозировку необходимо отменить ВАРГАТЕФ и проводить симптоматическую терапию.
Взаимодействие с другими лекарственными препаратами
Индукторы/ингибиторы P-gp
Нинтеданиб является субстратом для P-gp. Показано, что совместное применение с активным ингибитором P-gp кетоконазолом увеличивает экспозицию нинтеданиба, судя по величине AUC, в 1,61 раза, а судя по величине Сmах, в 1,83 раза.ли эритромицин) в случае совместного применения с препаратом ВАРГАТЕФ могут увеличивать экспозицию нинтеданиба. У таких пациентов переносимость нинтеданиба должна тщательно мониторироваться.
Активные индукторы P-gp (например, рифампицин, карбамазепин, фенитоин и препараты зверобоя продырявленного) могут уменьшать экспозицию нинтеданиба. Рекомендуется подбор альтернативной сопутствующей терапии с отсутствием или минимальным индуцирующим действием на систему P-gp.
Изоферменты CYP
Изоферменты CYP принимают лишь небольшое участие в биотрансформации нинтеданиба. В доклинических исследованиях нинтеданиб и его метаболиты (свободный кислый метаболит нинтеданиба и его глюкуронид) не ингибировали и не индуцировали изоферменты CYP. Поэтому вероятность лекарственных взаимодействий с нинтеданибом, основанных на влиянии на изоферменты CYP, считается небольшой.
Одновременная терапия с пирфенидоном
Было проведено изучение совместного применения нинтеданиба и пирфенидона у пациентов с ИЛФ в рамках специального фармакокинетического исследования. Пациенты первой группы получали однократную дозу 150 мг нинтеданиба до начала постепенного повышения дозы пирфенидона до максимальной (по 801 мг три раза в сутки), и еще одну дозу нинтеданиба 150 мг по достижении равновесного состояния (в последний день). Пациенты второй группы получали пирфенидон в постоянной дозе 801 мг три раза в сутки. У них была проведена оценка ФК профиля до начала совместной терапии и через 7 дней совместного приема нинтеданиба в дозировке 150 мг два раза в сутки. В первой группе скорректированные соотношения среднегеометрических значений (90 % доверительный интервал (ДИ) составляли 93% (57-151%) и 96% (70-131%) для показателей Сmах и AUCo-tz нинтеданиба соответственно (n=12). Во второй группе скорректированные соотношения среднегеометрических значений (90 % ДИ) составляли 97% (86-110%) и 95% (86-106 %) для показателей Cmax,ss и AUCτ,ss пирфенидона соответственно (n=12).
На основании полученных результатов можно сделать вывод об отсутствии признаков значимого фармакокинетического взаимодействия между нинтеданибом и пирфенидоном при совместном применении.
Одновременное применение с другими препаратами
Одновременное применение нинтеданиба с доцетакселом (75 мг/м2) в существенной степени не изменяет фармакокинетику этих препаратов.
Одновременное применение нинтеданиба с бозентаном не изменяет фармакокинетику нинтеданиба.
Возможность взаимодействий нинтеданиба с гормональными контрацептивными средствами не изучалась.
Одновременный прием с пищей
ВАРГАТЕФ рекомендуется принимать одновременно с пищей (см. раздел «Фармакокинетика»).
Особые указания
Желудочно-кишечные расстройства
- диарея
Лечение диареи (адекватная гидратация и антидиарейные лекарственные средства, например, лоперамид) следует проводить при появлении первых признаков. В случае развития диареи может потребоваться прерывание лечения, снижение дозы или прекращение терапии.
- тошнота и рвота
В случае сохранения симптомов тошноты и рвоты, несмотря на симптоматическое лечение (включая противорвотную терапию), может потребоваться снижение дозы, прерывание лечения или прекращение терапии.
Диарея и рвота могут приводить к обезвоживанию с нарушением или без нарушения водно-электролитного баланса, что может приводить к нарушениям функции почек. В случае обезвоживания рекомендуется восстановление водно-электролитного баланса.
Если возникают существенные желудочно-кишечные нежелательные явления, то необходимо проведение мониторирования концентрации электролитов в крови.
Нейтропения и сепсис
Во время терапии НМРЛ препаратом ВАРГАТЕФ, в частности при комбинации его с доцетакселом, необходимо проводить мониторинг форменных элементов крови на предмет развития нейтропении и возможных последующих осложнений в виде сепсиса или фебрильной нейтропении. Частое мониторирование всех форменных элементов крови следует осуществлять в начале каждого курса лечения у пациентов, получающих лечение нинтеданибом в комбинации с доцетакселом, в период наименьших значений нейтрофилов, а также при наличии клинических показаний после окончания последнего курса терапии данной комбинацией.
Нарушения функции печени
Безопасность и эффективность препарата не изучались у пациентов с нарушениями функции печени средней (класс В по шкале Чайлд-Пью) или тяжелой (класс С по шкале Чайлд-Пью) степени тяжести. Поэтому у таких пациентов применение препарата ВАРГАТЕФ не рекомендуется.
НМРЛ
В связи с повышением концентрации препарата в крови риск развития нежелательных явлений может быть выше у пациентов с легкими нарушениями функции печени (класс А по шкале Чайлд-Пью).
При проведении терапии нинтеданибом наблюдались случаи лекарственного поражения печени. В пострегистрационном периоде были получены сообщения о тяжелых формах лекарственного поражения печени с летальным исходом.
После начала терапии рекомендуется тщательный контроль активности печеночных трансаминаз, щелочной фосфатазы и концентрации билирубина (периодически, во время начала комбинированной терапии с доцетакселом, т.е. в начале каждого лечебного курса). Если отмечается существенное повышение активности печеночных ферментов, может потребоваться прерывание лечения, уменьшение дозы или прекращение терапии препаратом ВАРГАТЕФ.
При начале комбинированного лечения препаратом ВАРГАТЕФ и доцетакселом должны определяться уровни трансаминаз, ЩФ и билирубина. Эти показатели следует мониторировать в зависимости от клинических показаний или периодически во время лечения, например, в фазе комбинации с доцетакселом в начале каждого курса терапии и ежемесячно, если прием препарата ВАРГАТЕФ продолжается в виде монотерапии после прекращения терапии доцетакселом.
Если выявляется соответствующее повышение ферментов печени, может потребоваться перерыв в лечении, снижение дозы или прекращение терапии препаратом ВАРГАТЕФ (см. раздел «Способ применения и дозы»/ Таблица 2). Следует проводить поиск альтернативных причин повышения активности ферментов печени и принятие соответствующих мер при необходимости.
В случае повышения активности ферментов печени (АСТ/АЛТ) более чем в 3 раза по сравнению с ВЕН в сочетании с повышением уровня билирубина в 2 и более раза по сравнению с ВЕН, и повышением ЩФ менее чем в 2 раза по сравнению с ВГН, терапию препаратом ВАРГАТЕФ следует прервать. Если не будет установлена альтернативная причина изменений этих лабораторных показателей, препарат ВАРГАТЕФ следует окончательно отменить.
Для пациентов женского пола и азиатского происхождения характерен более высокий риск повышения активности печеночных ферментов.
Увеличение экспозиции нинтеданиба характеризовалось линейной зависимостью от возраста пациента, а также было обратно пропорционально массе тела, что также может повлечь за собой более высокий риск повышения активности печеночных ферментов. У пациентов с этими факторами риска рекомендуется тщательный контроль показателей функции печени.
ИЛФ и ИЗЛ-ССД
В связи с повышением концентрации препарата в крови риск развития нежелательных явлений может быть выше у пациентов с легкими нарушениями функции печени (класс А по шкале Чайлд-Пью). Пациенты с легкими нарушениями функции печени (класс А по шкале Чайлд-Пью) должны получать сниженную дозу препарата ВАРГАТЕФ.
При проведении терапии нинтеданибом наблюдались случаи лекарственного поражения печени. В пострегистрационном периоде были получены сообщения о несерьезных и серьезных случаях лекарственного поражения печени, включая тяжелые формы лекарственного поражения печени с летальным исходом.
Большая часть нарушений со стороны печени наблюдаются в течение первых 3-х месяцев терапии. Таким образом, рекомендуется оценивать активность печеночных трансаминаз и концентрацию билирубина до начала терапии препаратом ВАРГАТЕФ регулярно в течение первых трех месяцев терапии, а затем периодически во время лечения (например, при каждом визите пациента) или по клиническим показаниям.
Повышение активности печеночных ферментов (АЛТ, ACT, ЩФ. гамма-глутамилтрансфераза (ГГТ)) и билирубина было в большинстве случаев обратимым при уменьшении дозы или прерывании терапии.
В случае повышения уровня трансаминаз (ACT или АЛТ) более чем в 3 раза выше ВГН рекомендовано уменьшить дозу или прервать терапию препаратом ВАРГАТЕФ и проводить мониторирование пациента. Как только показатели трансаминаз вернутся к исходному уровню, доза препарата ВАРГАТЕФ может быть вновь повышена до полной (150 мг два раза в день) или лечение может быть возобновлено в сниженной дозе (100 мг два раза в день), которая впоследствии может быть повышена до полной дозы. Если повышение каких-либо показателей функции печени сопровождается клиническими признаками или симптомами повреждения печени, например, желтухой, лечение препаратом следует окончательно прекратить. Следует провести поиск альтернативных причин повышения активности ферментов печени.
Для пациентов с низкой массой тела (<65 кг), лиц азиатского происхождения и пациентов женского пола характерен более высокий риск повышения активности печеночных ферментов.
Увеличение экспозиции нинтеданиба характеризовалось линейной зависимостью от возраста пациента, что также может повлечь за собой более высокий риск повышения активности печеночных ферментов.
У пациентов с этими факторами риска рекомендуется тщательный контроль показателей функции печени.
Риск развития кровотечений
Данных о применении препарата ВАРГАТЕФ у пациентов с наследственной предрасположенностью к кровотечениям (например, болезнь фон Виллебранда) не имеется.
НМРЛ
Пациенты с недавно отмечавшимся легочным кровотечением (> 2,5 мл крови), а также пациенты с центрально расположенными опухолями и рентгенологическими признаками локальной инвазии крупных сосудов или с рентгенологическими признаками наличия полостей или некротических изменений опухоли исключались из клинических исследований. Поэтому применение препарата ВАРГАТЕФ у таких пациентов не рекомендуется.
- метастазы в головной мозг
Увеличения частоты церебральных кровотечений у пациентов с ранее адекватно лечеными метастазами в головной мозг (стабильными в течение ≥ 4 недель до начала лечения препаратом ВАРГАТЕФ) не наблюдалось. Однако такие пациенты должны тщательно мониторироваться в отношении признаков и симптомов церебральных кровотечений.
Пациенты с активными метастазами в головной мозг исключались из клинических исследований, применение препарата ВАРГАТЕФ у них не рекомендуется.
- терапия антикоагулянтами
В клинические исследования включались пациенты, получавшие длительную терапию низкомолекулярными гепаринами в низких дозах или ацетилсалициловой кислотой в дозах ≤ 325 мг/сут. Увеличения частоты кровотечений у таких пациентов не наблюдалось. Данных о применении препарата ВАРГАТЕФ у пациентов, предварительно получавших лечение антикоагулянтами в более высоких дозах, не имеется. Пациентам, у которых во время лечения препаратом ВАРГАТЕФ развивались тромбоэмболические нарушения, в связи с чем требовалась терапия антикоагулянтами, разрешалось продолжить прием препарата ВАРГАТЕФ, при этом увеличения частоты кровотечений не отмечалось. У пациентов, принимающих такие антикоагулянты, как варфарин или фенпрокумон, необходимо регулярно проверять протромбиновое время, международное нормализованное отношение (МНО) или следить за клиническими признаками кровотечений.
ИЛФ и ИЗЛ-ССД
В клинические исследования не включались пациенты с известным риском развития кровотечений, включая пациентов с наследственной предрасположенностью к кровотечениям или пациентов, получающих антикоагулянтную терапию в высоких дозах. Следовательно, данной категории пациентов лечение препаратом ВАРГАТЕФ должно быть назначено только в том случае, если потенциальная польза проводимой терапии превышает потенциальный риск.
Артериальная тромбоэмболия
Необходимо соблюдать осторожность при лечении пациентов с высоким сердечнососудистым риском, включая ишемическую болезнь сердца. Следует рассмотреть возможность перерыва в лечении пациентов, у которых развились симптомы острой ишемии миокарда.
Венозная тромбоэмболия
НМРЛ
У пациентов, получающих ВАРГАТЕФ, отмечался повышенный риск венозных тромбоэмболий, в том числе тромбоза глубоких вен. Пациенты должны тщательно мониторироваться в отношении тромбоэмболических нарушений. Пациентам с опасными для жизни венозными тромбоэмболиями ВАРГАТЕФ необходимо отменить.
ИЛФ и ИЗЛ-ССД
В клинических исследованиях не наблюдалось повышенного риска развития венозных тромбоэмболических осложнений. Однако в связи с особенностями механизма действия нинтеданиба. у пациентов может отмечаться повышенный риск развития тромбоэмболических явлений.
Перфорации ЖКТ
Частота перфораций ЖКТ в отдельных лечебных группах в клинических исследованиях была сопоставима. Основываясь на механизме действия препарата ВАРГАТЕФ. у пациентов возможен повышенный риск перфораций ЖКТ. В пострегистрационном периоде были получены сообщения о случаях перфорации ЖКТ, некоторые из которых приводили к летальному исходу. Особое внимание должно уделяться лечению пациентов, которые ранее подвергались абдоминальным хирургическим вмешательствам, или перенесших перфорацию полого органа в недавнем анамнезе, а также пациентов с ИЛФ, имеющих в анамнезе пептическую язву, дивертикулез или получающих сопутствующую терапию глюкокортикостероидами или НПВП. В связи с этим ВАРГАТЕФ может применяться только как минимум через 4 недели после больших хирургических, включая абдоминальные, вмешательств. В случае возникновения гастроинтестинальной перфорации терапия препаратом ВАРГАТЕФ должна прекращаться.
Нарушение заживления ран
Нинтеданиб, в связи с особенностями механизма действия, может нарушать заживление ран. В клинических исследованиях увеличения частоты нарушений заживления ран не наблюдалось. Специальных исследований, в которых бы изучалось влияние нинтеданиба на заживление ран, не проводилось. Поэтому лечение должно начинаться или возобновляться (если осуществлялся перерыв в связи с хирургическим вмешательством) с учетом клинического мнения об адекватности заживления раны.
Особые группы пациентов
В исследовании LUME-Lung 1 у пациентов с массой тела менее 50 кг, принимавших нинтеданиб в сочетании с доцетакселом, наблюдалась более высокая частота развития серьезных нежелательных явлений, чем у пациентов с массой тела ≥ 50 кг, хотя число пациентов с массой тела менее 50 кг было небольшим. Поэтому рекомендуется тщательное наблюдение за пациентами с массой тела менее 50 кг.
Влияние препарата на способность управления транспортными средствами, механизмами
Исследований по влиянию препарата на способность управлять транспортными средствами и занятию другими потенциально опасными видами деятельности, требующими повышенной концентрации внимания и быстроты психомоторных реакций не проводилось.
Во время применения препарата ВАРГАТЕФ пациентам нужно рекомендовать соблюдать осторожность при управлении транспортными средствами или механизмами.
Форма выпуска
Капсулы 100 мг
По 10 капсул в А1/А1 блистер. 6 блистеров в пачку картонную с инструкцией по медицинскому применению.
или
По 10 капсул в А1/А1 блистер. 6 блистеров в пачку картонную с инструкцией по медицинскому применению. По 2 пачки картонные в пленку из полипропилена.
Капсулы 150 мг
По 10 капсул в А1/А1 блистер. 6 блистеров в пачку картонную с инструкцией по медицинскому применению.
Условия хранения
Хранить в оригинальной упаковке при температуре не выше 25°С.
Хранить в недоступном для детей месте.
Срок годности
3 года.
Не использовать после истечения срока годности.
Условия отпуска
Отпускается по рецепту.
Наименование и адрес юридического лица, на имя которого выдано регистрационное удостоверение
Берингер Ингельхайм Интернешнл ГмбХ
55216, Бингер Штрассе 173, Ингельхайм-на-Рейне, Германия
Наименование и адрес места осуществления производства лекарственного препарата
Производитель готовой лекарственной формы
Каталент Германия Эбербах ГмбХ
69412, Гаммельсбахер Штр. 2, Эбербах, Германия
Первичная, вторичная упаковка и выпускающий контроль качества
Берингер Ингельхайм Фарма ГмбХ и Ко.КГ
55216, Бингер Штрассе 173, Ингельхайм-на-Рейне, Германия
Получить дополнительную информацию о препарате, а также направить свои претензии и информацию о нежелательных явлениях можно по следующему адресу в России
ООО «Берингер Ингельхайм»
125171, Москва, Ленинградское шоссе, 16А, стр.З
Комментарии
ПРАКТИКА ПЕДИАТРА



