Тафинлар - инструкция по применению
Синонимы, аналоги СтатьиРегистрационный номер:
ЛП 002274-060417
Торговое наименование препарата:
Тафинлар®
Международное непатентованное наименование:
дабрафениб
Лекарственная форма
Капсулы
Состав
Каждая капсула содержит:
Действующее вещество: дабрафениба мезилат микронизированный (в пересчете на дабрафениб) -59,25/88,88 мг (50,00/75,00 мг);
вспомогательныевещества:целлюлоза микрокристаллическая - 118,50/177,70 мг, магния стеарат -1,80/2,70 мг, кремния диоксид коллоидный - 0,45/0,68 мг. Состав капсулы из гипромеллозы размер №2, Swedish Orange (Шведский оранжевый)/размер №1, Opaque Pink (Непрозрачный розовый): краситель железа оксид красный - 1,29/0,56%, титана диоксид - 0,53/1,78%, гипрмеллоза q.s. до ЮО/q.s. до 100 %; чернила S-1-17822/S-1-17823: шеллак 44,5/44,5%, краситель железа оксид черный 23,4/23,4 мг, прониленгликоль 2,0/2,0%, аммиак водный 1,0/1,0%, бутанол 16,64/2,24%, изоиропанол 12,48/26,88%.
ОПИСАНИЕ
Капсулы 50 мг
Твердые непрозрачные капсулы с корпусом и крышечкой темно-красного цвета: на крышечке капсулы черными чернилами нанесено «GS TEW». на корпусе капсулы — «50 mg». Содержимое капсул представляет собой порошок- от белого до почти белого цвета. Размер капсул № 2.
Капсулы, 75 мг
Твердые непрозрачные капсулы с корпусом и крышечкой темно-розового цвета: на крышечке капсулы черными чернилами нанесено «GS LНF» на корпусе капсулы —«75 mg». Содержимое капсул представляет собой порошок от белого до почти белого цвета. Размер капсул №1.
Фармакотерапевтическая группа
Противоопухолевое средство, протеинкиназы ингибитор.
Код АТХ: не определен.
Фармакологические свойства
Механизм действия
Монотерапия
Дабрафениб является мощным селективным конкурирующим с АТФ ингибитором RAF-киназ; значения ICso для изоферментов BRAFV600E, BRAFV600K и BRAFV600D, составляют 0,65 нмоль, 0,5 нмоль и 1,84 нмоль соответственно.
Онкогенные мутации гена BRAF ведут к конститутивной активации пути RAS/RAF/MEK/ERK и стимуляции роста опухолевых клеток. Мутации гена BRAF с высокой частотой выявляются при специфических новообразованиях, включая меланому (около 50% случаев). Наиболее распространенные мутации гена, V600E, а также V600K, составляют 95% от всех мутаций гена BRAF у пациентов с различными злокачественными новообразованиями. В редких случаях обнаруживаются другие мутации, такие как V600D, V600G и V600R. Дабрафениб также ингибирует изоферменты CRAF и BRAF «дикого» типа, со значением ICso 5,0 нмоль и 3,2 нмоль соответственно. Дабрафениб ингибирует рост клеток меланомы, несущих мутацию гена BRAF V600, как in vitro, так и in vivo.
Мутации гена BRAF наблюдаются в 2% случаев немелкоклеточного рака легкого, и наиболее часто встречаются при аденокарциномах. Мутация BRAF V600E составляет примерно половину всех мутаций данного гена при немелкоклеточном раке легкого, кроме которой встречаются также другие мутации BRAF V600, например V600K, которая также приводит к конститутивной активации BRAF и является чувствительной к ингибиторам BRAF.
Комбинация с траметинибом
Траметиниб является высокоселективным обратимым аллостерическим ингибитором активации митоген-активируемых киназ 1 (МЕК1) и 2 (МЕК2), регулируемых внеклеточным сигналом. Белки МЕК являются компонентами сигнального пути ERK (киназы, регулируемой внеклеточным сигналом).
При комбинированном применении дабрафениб и траметиниб ингибируют две киназы в данном сигнальном пути, BRAF и МЕК. Применение этих препаратов в комбинации позволяет
достичь двойного ингибирования проведения пролиферативного сигнала. Комбинированное применение дабрафениба и траметиниба оказывает синергическое действие в клеточных линиях меланомы с мутацией BRAF in vitro и задерживает развитие резистентности in vivo в ксенотрансплантатах меланомы, несущих мутацию BRAF V600.
Фармакодинамика
Дабрафениб подавляет нижележащий фармакодинамический маркер (фосфорилированную ERK) в клетках меланомы, несущих мутацию BRAF V600, как in vitro, так и у животных. У пациентов с меланомой с мутацией BRAF V600 дабрафениб подавляет активность фосфорилированной ERK-киназы по отношению к исходному уровню.
Фармакокинетика
Всасывание
Максимальная концентрация дабрафениба в плазме крови (Сmах) после приема внутрь достигается в среднем через 2 часа.
Средняя абсолютная биодоступность дабрафениба при приеме внутрь составляет 95%. Экспозиция дабрафениба (Сmax И площадь под фармакокинетической кривой «концентрация- время» (AUC)) увеличивается пропорционально дозе после однократного приема внутрь в диапазоне доз от 12 мг до
300 мг, однако при повторном приеме 2 раза в сутки увеличение экспозиции менее пропорционально дозе.
При многократном применении экспозиция дабрафениба несколько снижается, возможно за счет индукции собственного метаболизма. Среднее соотношение кумуляции AUC день 18 / день 1 составило 0,73. После приема дабрафениба в дозе 150 мг 2 раза в сутки среднее геометрическое Cmax, AUQo-t) и концентрация перед приемом дозы (Сt) составили 1478 нг/мл, 4341 нг х час/мл и 26 нг/мл соответственно.
При применении во время еды биодоступность дорафениба снижается (Сmax и AUC ) уменьшаются на 51 и 31% соответственно), всасывание замедляется по сравнению с приемом дабрафениба в капсулах натощак.
Распределение
Дабрафениб связывается с белками плазмы крови (степень связывания составляет 99,7%). Кажущийся объем распределения составляет 70,3 л. Объем распределения в равновесном состоянии после внутривенного введения микродозы составляет 46 л. Дабрафениб in vitro является субстратом Р-гликопротеина человека (Pgp) и BCRP1. Однако данные переносчики оказывают незначительное влияние на биодоступность и элиминацию при пероральном применении, поэтому риск лекарственных взаимодействий минимален.
Метаболизм
Первый этап метаболизма дабрафениба - образование гидроксидабрафениба, катализируемое изофсрментами CYP2C8 и CYP3A4. Затем гидроксидабрафениб окисляется до карбоксидабрафениба с помощью изофермента CYP3A4. Далее возможно неферментативное декарбоксилирование карбоксидабрафениба с образованием дезметилдабрафениба.
Карбоксидабрафениб выводится с желчью и мочой. Дезметилдабрафениб может также образовываться в кишечнике и реабсорбироваться. Дезметилдабрафениб окисляется изоферментом CYP3A4. Конечный период полувыведения гидроксидабрафениба соответствует периоду полувыведения исходного соединения (10 часов), тогда как карбокси- и дезметил- метаболиты дабрафениба характеризуются более длительным периодом полувыведения (21-22 часа). После повторного приема препарата соотношение средних AUC метаболита и исходного соединения составили 0,9, 11 и 0,7 для гидрокси-, карбокси- и дезметилдабрафениба соответственно. Исходя из экспозиции, относительной эффективности и фармакокинетических свойств, гидрокси- и дезметилдабрафениб, вероятно, имеют важное значение в реализации клинической эффективности дабрафениба; активность карбоксидабрафениба, вероятнее всего, значительной роли не играет.
Выведение
Конечный период полувыведения дабрафениба после внутривенного введения микродозы составляет 2,6 часа. Конечный период полувыведения дабрафениба после приема внутрь составляет 8 часов (в связи с удлинением терминальной фазы). Клиренс из плазмы крови после внутривенного введения составляет 12 л/ч. При приеме 2 раза в сутки клиренс дабрафениба составляет 17,0 л/ч после однократного применения и 34,4 л/ч через 2 недели. Основным путем после перорального применения является выведение через кишечник (71% дозы, меченой радиоактивной меткой), почками выводится лишь 23% дозы, меченой радиоактивной меткой.
Особые группы пациентов
Пациенты с нарушением функции печени
Фармакокинетика дабрафениба была описана в клинических исследованиях у 65 пациентов с нарушением функции печени легкой степени (согласно классификации Национального института рака, NCI) с помощью популяционного анализа. У данных пациентов клиренс дабрафениба после приема внутрь незначительно отличался от клиренса дабрафениба у лиц с нормальной функцией печени (различия 4%). Кроме того, нарушение функции печени легкой степени не оказывало значимого влияния на концентрацию метаболитов дабрафениба в плазме крови. Нет данных по применению препарата у пациентов с нарушением функции печени средней и тяжелой степени. Дабрафениб следует с осторожностью применять у пациентов с нарушением функции печени средней и тяжелой степени (см. раздел «Способ применения и дозы»).
Пациенты с нарушением функции почек
Фармакокинетика дабрафениба была описана в клинических исследованиях с помощью популяционного анализа у 233 пациентов с нарушением функции почек легкой степени (СКФ (скорость клубочковой фильтрации) 60-89 мл/минх 1,73 м2) и 30 пациентов с нарушением функции почек средней степени (СКФ 30-59 мл/мин* 1,73 м2). У данных пациентов нарушение функции почек оказывало слабое и клинически незначимое влияние на клиренс дабрафениба после приема внутрь (<6% у пациентов обеих категорий). Нарушение функции почек слабой или средней степени не оказывало влияния на концентрацию гидрокси-, карбокси- и дезметилдабрафениба в плазме крови. Данные о применении дабрафениба у пациентов с нарушением функции почек тяжелой степени отсутствуют (см. раздел «Способ применения и дозы»).
Дети
Применение дабрафениба у пациентов младше 18 лет не изучено (см. раздел «Противопоказания»).
Пациенты пожилого возраста
По данным популяционного фармакокинетического анализа возраст не оказывает значительного влияния на фармакокинетику дабрафениба. Возраст старше 75 лет является прогностическим параметром более высоких плазменных концентраций карбокси- и дезметилдабрафениба в плазме крови с 40% увеличением экспозиции по сравнениюс аналогичными показателями у пациентов моложе 75 лет.
Масса тела и пол
По данным популяционного фармакокинетического анализа, масса тела и пол оказывают влияние на клиренс дабрафениба при применении внутрь; масса тела также влияет на объем распределения после приема внутрь и клиренс распределения. Данные фармакокинетические различия не расцениваются как клинически значимые.
Расовая принадлежность
По данным популяционного фармакокинетического анализа у пациентов азиатской и европеоидной расы не отмечено значимых различий фармакокинегики дабрафениба.
Комбинация с траметинибом
Многократное повторное применение дабрафениба в дозе 150 мг 2 раза в сутки одновременно с траметинибом в дозе 2 мг один раз в сутки приводило к увеличению Стах и AUC дабрафениба на 16% и 23% соответственно. По данным популяционного фармакокинетического анализа рассчитано незначительное уменьшение биодоступности траметиниба, соответствующее уменьшению AUC на 12%.
Показания к применению
• Нерезектабельная или метастатическая меланома
Препарат Тафинлар® в монотерапии и в комбинации с траметинибом показан для лечения пациентов с нерезектабельной или метастической меланомои с мутацией гена BRAF V600.
• Распространенный немелкоклеточныи рак легкого
Препарат Тафинлар* в комбинации с траметинибом показан для лечения пациентов с распространенным немелкоклеточным раком легкого с мутацией гена BRAF V600.
Противопоказания
- Повышенная чувствительность к дабрафенибу или другим компонентам, входящих в состав препарата.
- Беременность и период грудного вскармливания.
- Детский возраст до 18 лет.
- Противопоказано применение у пациентов с меланомои или немелкоклеточным раком легкого с «диким» типом гена BRAF в связи с отсутствием данных по эффективности и безопасности.
Применение при беременности и в период грудного вскармливания
Применение препарата Тафинлар® во время беременности и в период грудного вскармливания противопоказано.
Беременность
Резюме рисков
В исследованиях у животных дабрафсниб проявлял эмбриотоксические и тератогенные свойства. У животных при применении дабрафениба отмечено отставание развития скелета и снижение массы тела при рождении при концентрации, в 0,5 раз превышающей таковую при применении у человека в максимальной рекомендованной дозе 150 мг 2 раза в сутки. При применении дабрафениба у животных при концентрациях, трижды превышающих таковую при применении у человека в максимальной рекомендованной дозе 150 мг 2 раза в сутки, также отмечена эмбриолетальность, дефекты межжелудочковой перегородки и вариации формы вилочковой железы. Следует проинформировать беременную пациентку о возможном риске для плода.
Период грудного вскармливания
Резюме рисков
Данных о влиянии дабрафениба на детей, получающихгрудное молоко, а также о влиянии на продукцию грудного молока, нет. Учитывая, что многие лекарственные препараты проникают в грудное молоко, нельзя исключить риск для детей, получающих грудное молоко. Следует предупредить кормящую мать о потенциальном риске для ребенка. Следует исходить из пользы грудного вскармливания для ребенка и важности лечения для матери с учетом потенциальных побочных явлений у ребенка, получающего грудного молоко.
Контрацепция
В исследованиях у животных сообщалось о репродуктивной токсичности дабрафениба. Пациенткам с сохраненным репродуктивным потенциалом следует использовать надежные методы контрацепции (при правильном и длительном использовании которых частота наступления беременности составляет <1%) во время лечения и в течение 4 недель после прекращения лечения дабрафенибом, а также в течение минимум 4 месяцев после последнего приема траметиниба. Одновременное применение дабрафениба с
гормональными контрацептивами может снизить эффективность последних, в связи с чем рекомендуется использовать альтернативные средства контрацепции, такие как барьерные методы (см. раздел «Взаимодействие лекарственными препаратами»). В случае применения дабрафениба во время беременности, а также при наступлении беременности в этот период пациентку следует проинформировать о потенциальном риске для плода.
Способ применения и дозы
Лечение препаратом Тафинлар должен проводить врач, имеющий опыт применения противоопухолевых препаратов. Препарат принимают внутрь.
Перед началом применения препарата Тафинлар® необходимо получить подтверждение мутации гена BRAF V600 с помощью одобренного или валидированного теста у каждого пациента. Эффективность и безопасность препарата Тафинлар® у пациентов с меланомой или немелкоклеточным раком легкого с геном BRAF «дикого» типа не установлена. Препарат Тафинлар* противопоказан для лечения пациентов с меланомой с геном BRAF «дикого» типа. При применении препарата Тафинлар® в комбинации с траметинибом, последний следует применять в дозах, указанных в разделе «Способ применения и дозы» инструкции но применению траметиниба.
Взрослые
Рекомендуемая доза, как в монотерапии, так и в комбинации с траметинибом, составляет 150 мг 2 раза в сутки (соответствует суммарной суточной дозе 300 мг). Капсулы препарата Тафинлар® следует принимать внутрь не позднее, чем за 1 час до приема пищи или не раньше, чем через 2 часа после приема пищи, соблюдая 12-часовой интервал между приемом каждой дозы. В случае пропуска приема очередной дозы препарата Тафинлар® пропущенную дозу принимать не следует, если до приема следующей дозы остается менее 6 часов. При применении препарата Тафинлар® в комбинации с траметинибом однократную суточную дозу траметиниба следует принимать каждый день в одно и то же время вместе с утренней или вечерней дозой препарата Тафинлар®.
Коррекция дозы
Применение в монотерапии и в комбинации с траметинибом
При развитии нежелательных реакций может потребоваться прерывание лечения, уменьшение дозы или прекращение лечения (см. таблицы 1 и 2). Не рекомендовано корректировать дозу или прерывать терапию препаратом при развитии таких нежелательных реакций, как плоскоклеточный рак кожи (ПКРК) или новый очаг первичной меланомы (см. раздел «Особые указания»).
Рекомендуемая схема уменьшения дозы и рекомендации по коррекции дозы представлены в таблице 1 и таблице 2 соответственно. Уменьшение дозы препарата Тафинлар® менее 50 мг 2 раза в сутки не рекомендовано.
Таблица 1. Рекомендуемая схема уменьшения дозы препарата Тафинлар®
|
Изменение дозы |
Доза/схема приема |
|
Начальная доза |
150 мг 2 раза в сутки |
|
Первое уменьшение дозы |
100 мг 2 раза в сутки |
|
Второе уменьшение дозы |
75 мг 2 раза в сутки |
|
Третье уменьшение дозы |
50 мг 2 раза в сутки |
Таблица 2. Схема коррекции дозы препарата Тафинлар
|
Степень тяжести нежелательных реакций (по шкале СТС-АЕ)* |
Коррекция дозы |
|
Степень 1 или степень 2 (переносимые) |
Продолжение лечения и контроль в соответствии с клиническими показаниями. |
|
Степень 2 (непереносимые) или степень 3 |
Перерыв в лечении пока степень тяжести токсических реакций не достигнет 0—1 степени, при возобновлении лечения — уменьшение дозы на один уровень. |
|
Степень 4 |
Отмена терапии или перерыв в лечении пока степень тяжести токсических реакций не достигнет 0-1 степени, при возобновлении лечения — уменьшение дозы на один уровень. |
* — степень тяжести нежелательных реакций оценивается по шкале Стандартных критериев оценки нежелательных реакций (СТС-АЕ), версия 4.0
После эффективной коррекции нежелательного явления возможно увеличение дозы, которое проводится поэтапно в порядке обратном, указанному в таблицах 1 и 2. Доза препарата Тафинлар® не должна превышать 150 мг 2 раза в сутки.
При возникновении токсических реакций на фоне терапии препаратом Тафинлар® в комбинации с траметинибом следует либо снизить дозу обоих препаратов, либо прервать или полностью прекратить лечение обоими препаратами, за исключением случаев, описанных ниже.
Коррекция дозы только препарата Тафинлар® на фоне применения в комбинации с траметинибом необходима в случае развития следующих состояний:
- лихорадка, - увеит.
Лихорадка
Лечение препаратом Тафинлар®, как в монотерапии, так и в комбинации с траметинибом следует прервать при повышении температуры тела >38,5 °С, лечение траметинибом следует продолжать в той же дозе. Следует начать применение жаропонижающих препаратов, таких как ибупрофен или ацетаминофен/парацетамол, и провести обследование пациента с целью выявления возможных признаков и симптомов инфекции (см. раздел «Особые указания»). Прием препарата Тафинлар® может быть возобновлен после нормализации температуры тела в сочетании с соответствующей профилактикой жаропонижающими препаратами. Возобновление лечения препаратом Тафинлар® рекомендовано в той же дозе. Дозу препарата Тафинлар® следует снизить на один уровень в случае повторного развития лихорадки и/или в случае, если лихорадка сопровождается другими тяжелыми симптомами, включая обезвоживание, артериальную гипотензию или почечную недостаточность. В случае неэффективности жаропонижающих препаратов следует рассмотреть возможность применения пероральных глюкокортикостероидов.
Увеит
В случае развития увеита коррекции дозы препарата Тафинлар® не требуется, если симптомы воспалительного процесса хорошо контролируется лечением препаратами для местного применения. При отсутствии ответа на лечение увеита офтальмологическими препаратами для местного применения следует прервать прием препарата Тафинлар® с последующим возобновлением после купирования местного воспалительного процесса. Возобновлять прием препарата Тафинлар® следует в дозе, уменьшенной на один уровень. При применении препарата Тафинлар® в комбинации с траметинибом модификации дозы траметиниба не требуется. Примечание: перед применением траметиниба следует ознакомиться с разделом «Способ применения и дозы» инструкции но применению траметиниба.
Особые группы пациентов
Дети и подростки
Эффективность и безопасность препарата Тафинлар® у детей и подростков (младше 18 лет) не установлена (см. раздел «Противопоказания »).
Пациенты пожилого возраста
У пациентов старше 65 лет коррекции дозы не требуется (см. подраздел «Фармакокинетика»).
Пациенты с нарушением функции почек
У пациентов с нарушением функции почек легкой и средней степени коррекции дозы не требуется. По данным исследований не выявлено значимого влияния нарушения функции почек легкой и средней степени на клиренс дабрафениба и концентрацию его метаболитов после приема внутрь (см. подраздел «Фармакокинетика»). В связи с отсутствием клинических данных по применению препарата Тафинлар® у пациентов с нарушением функции почек тяжелой степени необходимость коррекции дозы препарата у пациентов данной категории не установлена. Следует с осторожностью проводить лечение препаратом у пациентов данной категории.
Пациенты с нарушением функции печени
У пациентов с нарушением функции печени легкой степени коррекции дозы препарата Тафинлар® не требуется. По данным исследований не выявлено значимого влияния нарушения функции печени легкой степени на клиренс дабрафениба и концентрацию его метаболитов после приема внутрь (см. подраздел «Фармакокинетика»). В связи с отсутствием клинических данных по применению препарата Тафинлар® у пациентов с нарушением функции печени средней и тяжелой степени необходимость коррекции дозы препарата у пациентов данной категории не установлена. Поскольку главными путями выведения дабрафениба являются метаболизм в печени с последующим выведением с желчью, у пациентов с нарушением функции печени средней и тяжелой степени повышен риск увеличения экспозиции дабрафениба. В связи с вышесказанным следует с осторожностью проводить лечение препаратом у пациентов данной категории.
Побочное действие
Данные клинических исследований
Нерезектабельная или метастатическая меланома
Монотерапия дабрафенибом
Объединенные данные по безопасности монотерапии препаратом Тафинлар® получены в результате пяти клинических исследований с участием 578 пациентов с меланомой, около 30% из которых получали дабрафениб более 6 месяцев.
В общей популяции для оценки безопасности дабрафениба наиболее частыми (>15%) нежелательными реакциями (HP) являлись гиперкератоз, головная боль, лихорадка, артралгия, утомляемость, тошнота, кожные папилломы, алопеция, кожная сыпь и рвота. HP представленные ниже, сгруппированы в соответствии с классификацией органов и систем органов MedDRA. В пределах каждого системно-органного класса HP распределены по частоте возникновения в порядке уменьшения их значимости.
Для оценки частоты встречаемости использованы следующие критерии: очень часто (>1/10), часто (>1/100 и <1/10), нечасто (>1/1000 и <1/100),редко (> 1/10000 и <1/1000), очень редко (<1/10000).
|
Доброкачественные, злокачественные и неуточненные новообразования (включая кисты и полипы) |
|
|
Очень часто: |
папиллома. |
|
Часто: |
акрохордон (мягкая бородавка), плоскоклеточный рак кожи, включая собственно ПРК, рак in situ (болезнь Боуэна) и кератоакантома, себорейный кератоз. |
|
Нечасто: |
новый очаг первичной меланомы. |
|
Нарушения со стороны иммунной системы |
|
|
Нечасто: |
гиперчувствительность. |
|
Инфекционные и паразитарные заболевания |
|
|
Часто: |
назофарингит. |
|
Нарушения со стороны обмена веществ и питания |
|
|
Очень часто: |
снижение аппетита. |
|
Часто: |
гипофосфатемия, гипергликемия. |
|
Нарушения со стороны нервной системы |
|
|
Очень часто: |
головная боль. |
|
Нарушения со стороны органа зрения |
|
|
Нечасто: |
увеит. |
|
Нарушения со стороны дыхательной системы, органов грудной клетки и средостения |
|
|
Очень часто: |
кашель. |
|
Нарушения со стороны желудочно-кишечного тракта |
|
|
Очень часто: |
тошнота, рвота, диарея. |
|
Чacто: |
запор. |
|
Нечасто: |
панкреатит. |
|
Нарушения со стороны колеи и подкожных тканей |
|
|
Очень часто: |
проявления со стороны кожи (сыпь, гиперкератоз), алопеция, синдром ладонно-подошвенной эритродизестезии. |
|
Часто: |
проявления со стороны кожи (актинический кератоз, повреждения кожи, сухость кожи, эритема, зуд). |
|
Нарушения со стороны скелетно-мышечной и соединительной ткани |
|
|
Очень часто: |
артралгия, миалгия, боль в конечностях, боль в спине. |
|
Нарушения со стороны почек и мочевыводящих путей |
|
|
Нечасто: |
почечная недостаточность, острая почечная недостаточность. |
|
Редко: |
тубулоинтерстициальный нефрит. |
|
Общие расстройства и нарушения в месте введения |
|
|
Очень часто: |
астения, озноб, утомляемость, лихорадка. |
|
Часто: |
гриппоподобный синдром. |
|
Лабораторные и инструментальные данные |
|
|
Очень часто: |
повышение активности щелочной фосфатазы. |
|
Часто: |
гипонатриемия. |
Комбинированная терапия
Оценку безопасности комбинированной терапии препаратом Тафинлар® с траметинибом проводили в двух клинических исследованиях с участием пациентов с нерезектабельной или метастатической меланомой с мутацией гена BRAF, которые принимали внутрь дабрафениб в дозе 150 мг 2 раза в сутки и траметиниб в дозе 2 мг 1 раз в сутки. Наиболее частыми HP (>20%) при применении дабрафениба в комбинации с траметинибом являлись лихорадка, утомляемость, тошнота, головная боль, озноб, диарея, сыпь, артралгия, артериальная гипертензия. рвота, периферические отеки и кашель. HP, зарегистрированные при проведении комбинированной терапии дабрафенибом и траметинибом в рамках клинических исследований приведены ниже.
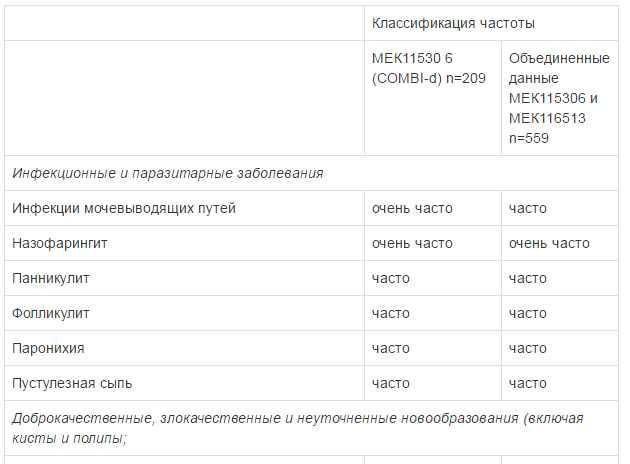
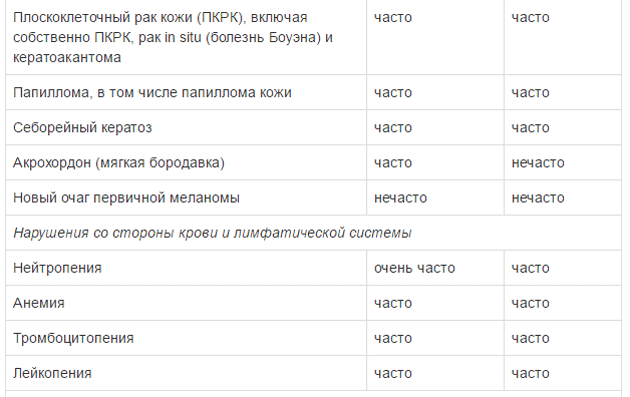
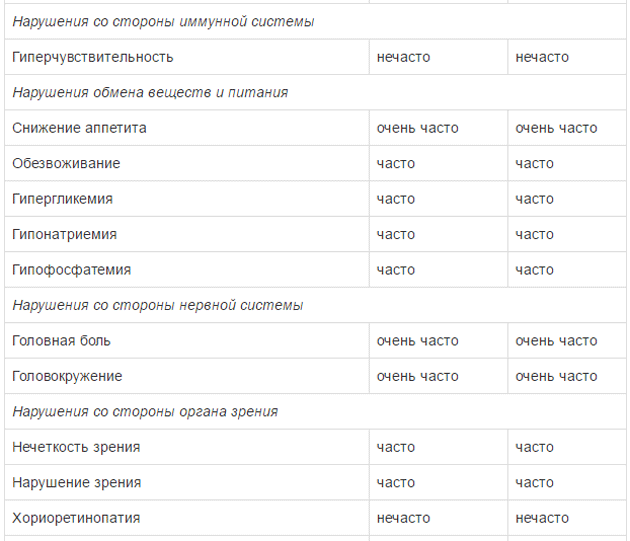
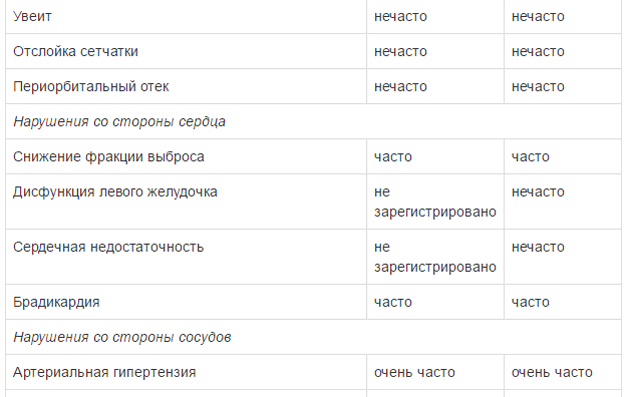
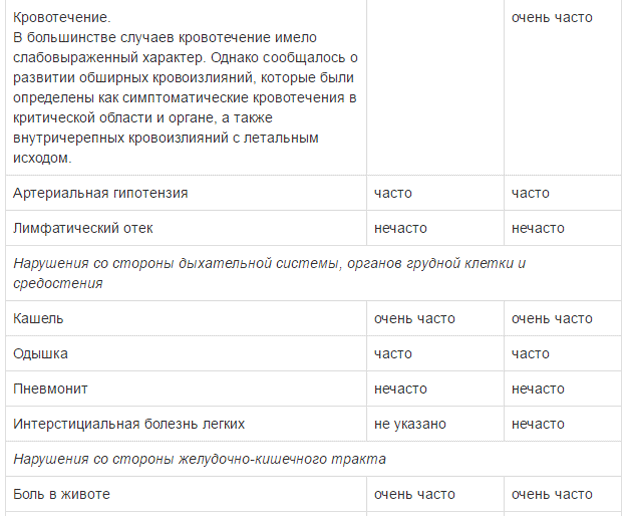
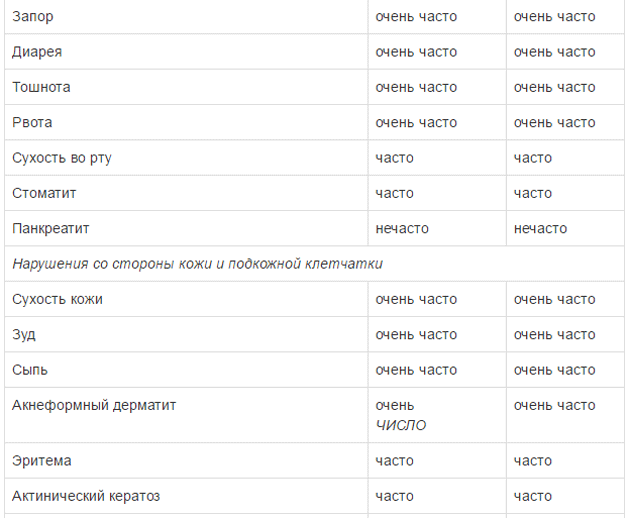
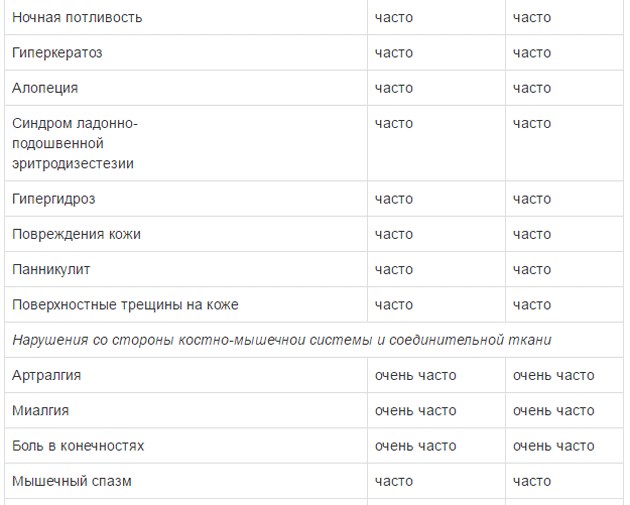
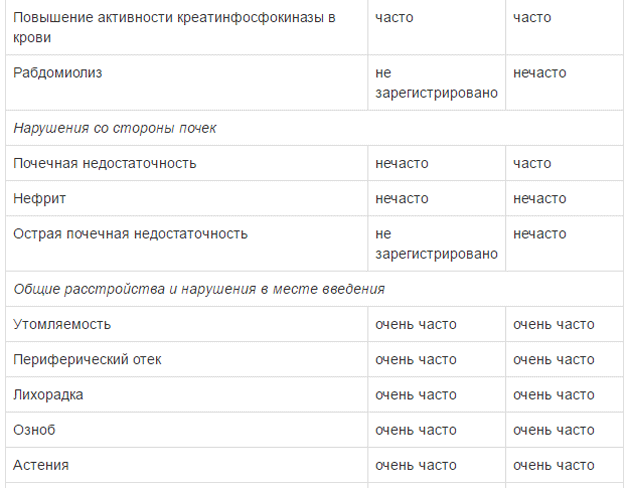
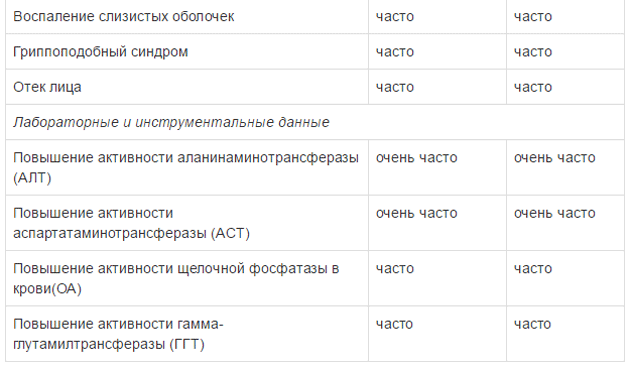
Удлинение интервала QT
В объединенной популяции у одного пациента была зарегистрирована длина интервала QTcB >500 мсек, у 3% пациентов максимальное зарегистрированное удлинение интервала QTc превышало 60 мсек.
Снижение ФВЛЖ (фракции выброса левого желудочка)
Уменьшение ФВЛЖ было зарегистрировано у 1% пациентов, при этом большинство случаев были бессимптомными и обратимыми. Нет данных о применении препарата Тафинлар у пациентов с показателем ФВЛЖ ниже нижней границы нормы.
Распространенный немелкоклеточный рак легкого
Монотерапия препаратом Тафинлар®
Безопасность монотерапии препаратом Тафинлар оценивалась в ходе клинического исследования у пациентов с метастатическим немелкоклеточным раком легкого с мутацией гена BRAF V600E. При применении препарата в дозе 150 мг два раза в день наиболее частыми HP были: лихорадка, астения, повышенная утомляемость, гиперкератоз, кашель, папиллома кожи, сухость кожных покровов, синдром ладонно-подошвенной эритродизестезии, алопеция, тошнота и одышка.
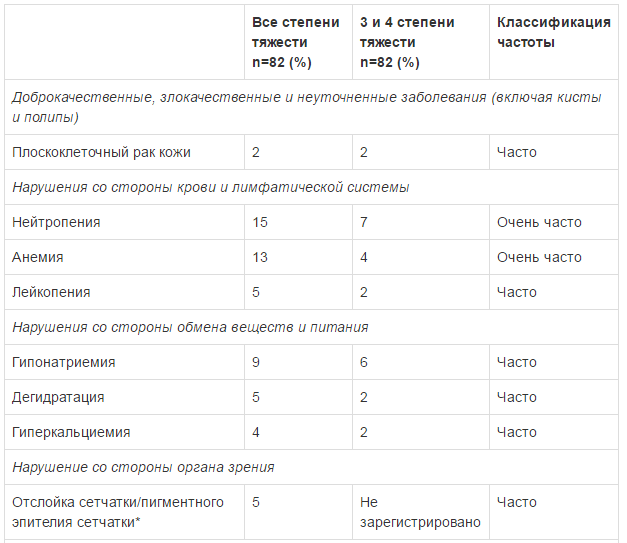
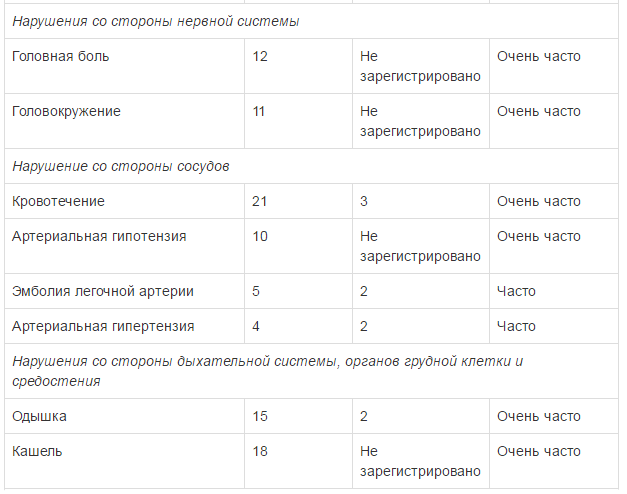
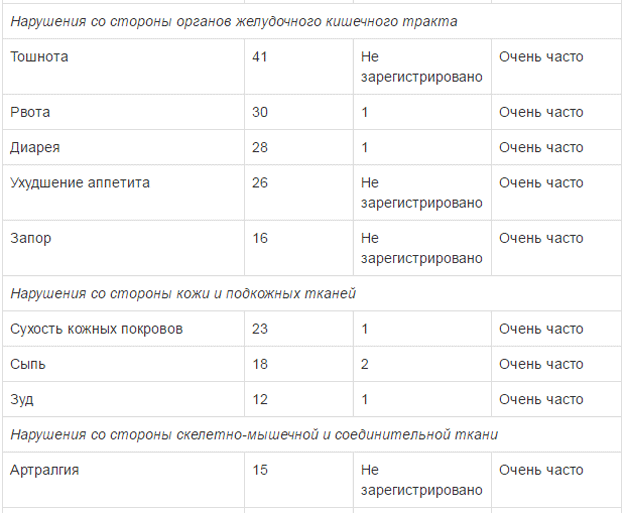
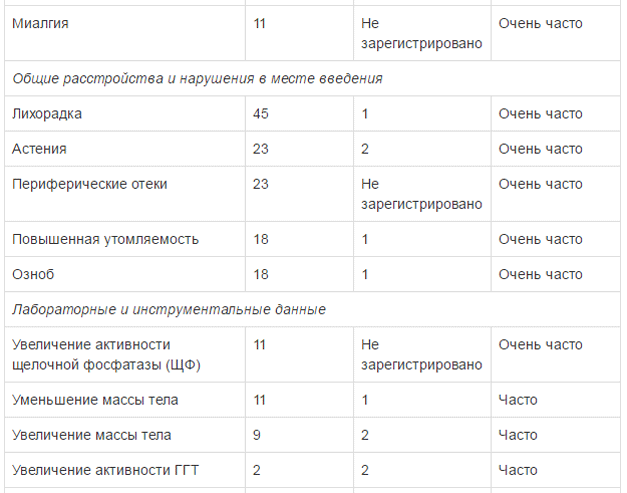
Безопасность применения препарата Тафинлар в комбинации с траметииибом оценивалась в ходе клинического исследования у пациентов с метастатическим немелкоклеточным раком легкого с мутацией гена BRAF V600E. Наиболее частыми HP (>20%) при применении комбинированной терапии были: лихорадка, тошнота, рвота, диарея, ухудшение аппетита, астения, сухость кожных покровов и периферические отеки. В таблице ниже указаны HP, возникавшие с частотой >10% (все стеиени тяжести) с учетом HP 3 и 4 степени тяжести, возникавших с частотой >2%, зарегистрированные в клиническом исследовании при применении препарата Тафинлар в комбинации с траметинибом у пациентов с немелкоклеточным раком легкого.

Необходимо соблюдать осторожность при применении мощных ингибиторов (например, кетоконазол, нефазодон, кларитромицин, ритонавир, гемфиброзил, или индукторов (например, рифампицин, фенитоин, карбамазепин, фенобарбитал, зверобой продырявленный) изоферментов CYP2C8 или CYP3A4 одновременно с дабрафенибом. Влияние дабрафениба на вещества транспортных систем In vitro дабрафениб является ингибитором белков-переносчиков органических анионов ОАТР1В1 и ОАТР1ВЗ, клиническую значимость данного явления нельзя исключить. По этой причине следует соблюдать осторожность при одновременном применении дабрафениба и субстратов ОАТВ1В1 или ОАТР1ВЗ, таких как статины.
Несмотря на то, что in vitro дабрафениб и его метаболиты (гидроксидабрафениб, карбоксидабрафениб и дезметилдабрафениб) проявляли свойства ингибиторов белков-переносчиков органических анионов ОАТ1 и ОАТЗ, данные клинических исследований позволяют предположить, что риск лекарственного взаимодействия минимален. Также было показано, что дабрафениб и дезметилдабрафениб являются умеренными ингибиторами белка резистентности рака молочной железы человека, однако, учитывая клиническую экспозицию, риск возникновения лекарственного взаимодействия минимален.
Препараты, которые влияют на рН желудочного сока
Препараты, которые изменяют рН верхнего отдела желудочно-кишечного тракта (например, ингибиторы протонной помпы, антагонисты Н2-рецепторов, антациды) могут изменять растворимость дабрафениба и снижать его биодоступность, однако, клинических исследований по оценке влияния препаратов, изменяющих рН желудочного сока, на системную экспозицию дабрафениба не проводилось. Исходя из вышесказанного, при одновременном применении дабрафениба с ингибиторами протонной помпы,антагонистами Н2-рецепторов или антацидами, системная экспозиция дабрафениба может снижаться, влияние данного явления на эффективность дабрафениба не установлена.
Влияние дабрафениба на другие лекарственные препараты
Дабрафениб усиливает CYP3A4- и СУР2С9-опосредованный метаболизм и может увеличить активность других
изоферментов системы цитохрома, включая изоферменты CYP2B6, CYP2C8 и CYP2C19 и УДФ-глюкуронозилтрансферазу (УГТ), а также может увеличить активность белков переносчиков (например, P-gp)- В клиническом исследовании у 12 пациентов, получивших однократную дозу мидозолама (субстрата изофермента CYP3A4) отмечено уменьшение его Сmах и AUC на 61% и 74% соответственно при повторном применении дабрафениба в дозе 150 мг 2 раза в сутки. В отдельном исследовании у 14 пациентов отмечено уменьшение AUC S-варфарина (субстрата изофермента CYP2C9) и R-варфарина (субстрата изоферментов CYP3A4/CYP1A2) на 37% и 33% соответственно после одновременного повторного применения дабрафениба. Ни дабрафениб, ни три его метаболита не оказывают ингибирующего действия на Pgp in vitro. Одновременное применение дабрафениба с лекарственными препаратами, чувствительными к индукции изоферментов CYP3A4 или CYP2C9 (например, гормональные контрацептивы, варфарин или дексаметазон), может привести к снижению их концентрации и потере эффективности. В случае, когда применение таких препаратов с дабрафенибом необходимо, следует контролировать состояние пациента для выявления потери эффективности данных препаратов или рассмотреть возможность альтернативной лекарственной терапии.
Ожидается большое количество препаратов, с которыми возможно развитие лекарственного взаимодействия при одновременном применении с дабрафенибом, однако интенсивность взаимодействия может быть различной. Группы вышеописанных лекарственных препаратов могут включать в т.ч.:
- анальгетики (например, фентанил, метадон);
- антибиотики (например, кларитромицин, доксициклин);
- противоопухолевые препараты (например, кабазитаксел);
- антикоагулянты (например, аценокумарол, варфарин);
- противоэпилептические препараты (например, карбамазепин, фенитоин, примидон, вальпроевая кислота);
- нейролептики (например, галоперидол);
- блокаторы кальциевых каналов (например, дилтиазем, фелодипин, никардипин, верапамил);
- сердечные гликозиды (например, дигоксин);
глюкокортикостероиды (например, дексаметазон, метилпреднизолон);
- противовирусные препараты для лечения ВИЧ-инфекции (например, ампренавир, атазанавир, дарунавир, делавирдин, эфавиренз, фосампрснавир, индинавир, лопинавир, нелфинавир, саквинавир, типранавир);
- гормональные контрацептивы;
- снотворные препараты (например, диазепам, мидазолам, золпидем);
- иммунодепрессанты (например, циклоспорин, такролимус, сиролимус);
статины, метаболизируемые изоферментом CYP3A4 (например, аторвастатин, симвастатин).
Применение в комбинации с траметинибом
Многократное одновременное применение дабрафениба в дозе 150 мг 2 раза в сутки и траметиниба в дозе 2 мг 1 раз в сутки приводило к увеличению Сmах и AUC дабрафениба на 16% и 23% соответственно. Информация о взаимодействии траметиниба с другими лекарственными препаратами указана в инструкции по применению траметиниба. По данным популяционного фармакокинетнческого анализа при применении траметиниба с дабрафенибом отмечается небольшое уменьшение биодоступности траметиниба, соответствующее уменьшению AUC последнего на 12%. Указанные изменения Сшах и AUC дабрафениба и траметиниба не являются клинически значимыми.
Особые указания
Перед началом применения препарата Тафинлар® в комбинации с траметинибом ознакомьтесь с полной инструкцией по медицинскому применению указанного лекарственного препарата, в т.ч. с разделом «Особые указания».
Лихорадка
У пациентов, получавших препарат Тафинлар® как в монотерапии, так и в составе комбинированной терапии с траметинибом, отмечалось появление лихорадки. Тяжесть и частота данного явления увеличивалась при применении препарата в составе комбинированной терапии с траметинибом по поводу нерезектабельной или метастатической меланомы; при терапии препаратом Тафинлар® в комбинации с траметинибом по поводу немелкоклеточного рака легкого тяжесть и частота развития лихорадки увеличивалась в меньшей степени.
При применении препарата Тафинлар® в дозе 150 мг 2 раза в сутки в комбинации с траметинибом в дозе 2 мг 1 раз в сутки у пациентов с меланомой в половине случаев первые эпизоды лихорадки отмечались в первый месяц терапии, при этом приблизительно у 1/3 пациентов, получавших комбинированную терапию, отмечалось 3 или более эпизодов лихорадки. Данное состояние может сопровождаться выраженной дрожью, обезвоживанием и артериальной гипотензией, в редких случаях возможно развитие острой почечной недостаточности. Во время и после тяжелых эпизодов лихорадки следует контролировать сывороточную концентрацию креатинина и другие показатели функции почек. Отмечены случаи тяжелой неинфекционной фебрильной лихорадки. При применении препарата в клинических исследованиях данные состояния успешно купировались коррекцией дозы препарата Тафинлар® и/или приостановлением лечения с применением поддерживающей терапии (см. раздел «Способ применения и дозы»). Для коррекции дозы траметиниба ознакомьтесь с инструкцией по медицинскому применению вышеуказанного лекарственного препарата, в т.ч. с разделом «Способ применения и дозы».
Плоскоклеточный рак кожи (ПКРК)
У пациентов, получавших препарат Тафинлар®, как в монотерапии, так и в составе комбинированной терапии с траметинибом, сообщалось о случаях ПКРК (в т.ч. классифицированных как кератоакантома и смешанная кератоакантома). При монотерапии препаратом Тафинлар® возникновение ПКРК отмечено у 10% пациентов, при этом медиана времени до возникновения первого проявления ПКРК составила 8 недель. При комбинированной терапии препаратом Тафинлар® с траметинибом возникновение ПКРК отмечено у 3% пациентов, при этом медиана времени до возникновения первого проявления ПКРК составила от 20 до 32 недель. Более чем у 90% пациентов с ПКРК, возникшем на фоне применения препарата Тафинлар®, лечение препаратом было продолжено без коррекции дозы.
В клиническом исследовании развитие ПКРК отмечено у 18% пациентов с немелкоклеточным раком легкого, получавших монотерапию препаратом Тафинлар®, при этом медиана времени до возникновения первого проявления составила 11 недель. У пациентов, получавших терапию препаратом в комбинации с траметинибом возникновение ПКРК отмечено у 2% пациентов.
Следует оценить состояние кожных покровов перед началом лечения препаратом Тафинлар* с последующим контролем каждые 2 месяца на протяжении всего курса лечения. Следует контролировать состояния кожных покровов каждые 2— 3 месяца на протяжении 6 месяцев после окончания терапии препаратом или до начала другой противоопухолевой терапии. При подтверждении диагноза ПКРК следует провести хирургическое лечение пораженного участка, терапию препаратом Тафинлар® следует продолжить без коррекции дозы. Следует проинструктировать пациента о необходимости сообщать лечащему врачу о возникновении новых очагов поражения на коже. Новые очаги первичной меланомы
Сообщалось о возникновении новых очагов первичной меланомы у пациентов, принимавших препарат Тафинлар® по поводу нерезектабельной или метастатической меланомы. В клинических исследованиях данные случаи выявлялись в течение первых 5 месяцев терапии и не требовали коррекции лечения, кроме резекции пораженного участка. Необходим регулярный контроль состояния кожных покровов (по схеме, описанной для ПКРК).
Вторичные/рецидивирующие злокачественные образования некожной локализации
In vitro наблюдалась парадоксальная активация МАР-киназного сигнального каскада в клетках с «диким» типом BRAF с RAS мутацией, подвергавшихся воздействию ингибиторов BRAF, что может приводить к увеличению риска развития злокачественных образований иной локализации у пациентов, получающих препарат Тафинлар®. При применении ингибиторов BRAF были зарегистрированы случаи развития злокачественных образований, несущих мутацию RAS. Пациентам, получающим терапию препаратом, следует обеспечить соответствующее наблюдение. При выявлении злокачественного новообразования иной локализации, несущего RAS-мутацию, у пациента, получающего терапию препаратом Тафинлар®, лечение препаратом следует продолжить, исходя из соотношения польза/риск. При применении препарата Тафинлар® в комбинации с траметинибом коррекции дозы последнего не требуется. Следует продолжить контроль состояния пациента с целью выявления вторичных/рецидивирующих RAS-положительных злокачественных новообразований иной локализации на период до 6 месяцев после отмены терапии препаратом Тафинлар® или вплоть до начала иной противоопухолевой терапии.
Панкреатит
Менее чем у 1 % пациентов, получавших препарат Тафинлар® в клинических исследованиях по поводу меланомы, отмечено развитие панкреатита, данное состояние также зарегистрировано в клинических исследованиях у 4% пациентов, получавших препарат Тафинлар* по поводу немелкоклеточного рака легкого.
В одном из случаев состояние возникло у пациента с меланомой при применении первой дозы препарата с повторным возникновением при повторном применении препарата Тафинлар® в уменьшенной дозе. Необходимо немедленно обследовать пациентов с жалобами на боль в животе неясной этиологии с определением активности амилазы и липазы в сыворотке крови. Следует тщательно наблюдать пациента при возобновлении лечения дабрафенибом после эпизода панкреатита.
Препарат Тафинлар® в комбинации с траметинибом: см. полную инструкцию по применению траметиниба, раздел «Особые указания». Кровотечение
На фоне приема препарата Тафинлар® как в монотерапии, так и в комбинации с траметинибом зарегистрированы серьезные случаи геморрагических явлений (см. раздел «Побочное действие»). В клинических исследованиях у 6 пациентов из 559 (1%), получавших препарат Тафинлар® в комбинации с траметинибом по поводу нерезектабельной или метастатическоймеланомы, развилось внутричерепное кровоизлияние с летальным исходом; в клиническом исследовании при применении препарата Тафинлар® в комбинации с траметинибом по поводу распространенного немелкоклеточного рака легкого, внутричерепное кровоизлияние с легальным исходом зарегистрировано у 2 пациентов из 82 (2%). При появлении симптомов кровотечения пациенту следует немедленно обратиться за медицинской помощью. Удлинение интервала QT
Максимальное зарегистрированное удлинение интервала QTc, превышающее 60 мсек, наблюдалось у 3% пациентов, получавших лечение дабрафенибом (отмечен один случай удлинения интервала QTc >500 мсек). Не рекомендовано применение препарата Тафинлар® у пациентов с нарушением водно-электролитного баланса (в т.ч. содержания магния), не поддающегося коррекции, синдромом удлинения интервала QT, а также у пациентов, получающих препараты, способные удлинять интервал QT. Следует провести электрокардиографическое исследование (ЭКГ), а также определить содержание электролитов (в том числе, магния) перед началом терапии препаратом, а также через 1 месяц после ее начала и после любой коррекции дозы. В дальнейшем контроль рекомендован, в частности, у пациентов с нарушением функции печени средней или тяжелой степени ежемесячно на протяжении первых 3 месяцев лечения, а затем каждые 3 месяца или чаще, в соответствии с клиническими показаниями. У пациентов с длиной интервала QTc >500 мсек не следует начинать терапию препаратом Тафинлар®. При увеличении интервала QTc >500 мсек на фоне терапии препаратом следует временно приостановить лечение, провести коррекцию водно-электролитного баланса (в т.ч. содержания магния) и скорректировать факторы риска удлинения интервала QT со стороны сердца (например, застойная сердечная недостаточность, брадикардия). Лечение следует возобновить в сниженной дозе (см. таблицу 2) после укорочения интервала QTc до значения <500 мсек. Следует полностью прекратить лечение препаратом при увеличении интервала QTc >500 мсек с увеличением более чем на 60 мсек от исходного значения.
Гипергликемия
При применении препарата Тафинлар® отмечалось развитие гипергликемии. В клиническом исследовании у 5 из 12 пациентов с сахарным диабетом в анамнезе при применении дабрафениба возникла необходимость в увеличении интенсивности гипогликемической терапии. Частота развития гипергликемии 3 степени по данным лабораторных исследований составила 6% (12/187) у пациентов, получавших дабрафениб, в отличие от пациентов, получавших дакарбазин, у которых такие случаи не зарегистрированы. При лечении препаратом Тафинлар® у пациентов с ранее диагностированным сахарным диабетом или гипергликемией необходимо проводить рутинный контроль концентрации глюкозы в сыворотке крови. Пациентам рекомендуется сообщать лечащему врачу о симптомах тяжелой гипергликемии, таких как чрезмерная жажда или увеличение объема и частоты мочеиспусканий. Дефицит глюкозо-6-фосфат-дегидрогеназы Дабрафениб, содержащий сульфонамидную группу,увеличивает риск развития гемолитической анемии у пациентов с дефицитом глюкозо-6-фосфатдегидрогеназы (Г6ФДГ). Необходимо проводить тщательный медицинский контроль состояния пациентов с дефицитом Г6ФДГ на предмет признаков гемолитической анемии.
ФОРМА ВЫПУСКА
Срок годности
применять после истечения срока годности, указанного на упаковке.Условия хранения
Хранить при температуре не выше 30°С.
Хранить в недоступном для детей месте.
Условия отпуска
По рецепту
Производитель
Производитель готовой лекарственной формы
ГлаксоСмитКляйн Инк., 7333 Миссисога Роад, Миссисога,
Онтарио, Канада, L5N 6L4 /
GlaxoSmithKline Inc., 7333, Mississauga Rd.,
Mississauga, Ontario, Canada, L5N 6L4
Глаксо Оперэйшенс Великобритания Лимитед,
Прайори стрит, Вэа, Хертфордшир, SG12 0DJ, Великобритания /
Glaxo Operations UK Limited,
Priory Street, Ware, Hertfordshire, SGI2 ODJ, United Kingdom
Фасовщик (первичная) упаковка
Глаксо Вэллком С.А., Авда. де Экстремадура 3, 09400-Аранда де Дуэро, Бургос, Испания /
Glaxo Wellcome S.A., Avda. de Extremadura 3, 09400-Aranda de Duero, Burgos, Spain
Упаковщик (вторичная (потребительская) упаковка), выпускающий контроль качества
Глаксо Вэллком С.А., Авда. де Экстремадура 3, 09400-Аранда де Дуэро,Бургос, Испания /
Glaxo Wellcome S.A.,Avda. de Extremadura 3, 09400-Aranda de Duero, Burgos, Spain
391800, Россия, Рязанская обл., Скопинский район, с. Успенское.
Комментарии
ПРАКТИКА ПЕДИАТРА



