Эмплисити - инструкция по применению
Синонимы, аналоги СтатьиРЕГИСТРАЦИОННЫЙ НОМЕР:
ЛП-004241 - 120417
ТОРГОВОЕ НАЗВАНИЕ:
Эмплисити® (Empliciti®)
МЕЖДУНАРОДНОЕ НЕПАТЕНТОВАННОЕ НАЗВАНИЕ:
элотузумаб
ЛЕКАРСТВЕННАЯ ФОРМА:
лиофилизат для приготовления концентрата для приготовления раствора для инфузий
СОСТАВ:
1 флакон содержит*:
активное вещество: элотузумаб 340 мг или 440 мг,
вспомогательные вещества: натрия цитрата дигидрат 16,6 мг или 21,5 мг; лимонная кислота моногидрат 2,44 мг или 3,17 мг; сахароза 510 мг или 660 мг; полисорбат 80 3,40 мг или 4,40 мг.
* Фасовка производится с учетом перезакладки в 40 мг (13,3%) для 300 мг и 40 мг (10%) для 400 мг, что необходимо для гарантии полного извлечения заявленной дозировки. Извлекаемое количество элотузумаба в одном флаконе - 300 мг и 400 мг соответственно.
ОПИСАНИЕ
Пористая масса или порошок от белого до почти белого цвета.
ФАРМАКОТЕРАПЕВТИЧЕСКАЯ ГРУППА:
противоопухолевое средство, антитела моноклональные
Код ATX: L01XC23
ФАРМАКОЛОГИЧЕСКОЕ ДЕЙСТВИЕ
Фармакодинамика
Элотузумаб является человеческим иммуностимулирующим моноклональным IgGl антителом, которые специфически связывается с белком SLAMF7 (7 представитель в семействе сигнальных молекул активации лимфоцитов). SLAMF7 в большом количестве экснрессируется на миеломных клетках независимо от типа цитогенетических аномалий. SLAMF7 также экспрессируется на натуральных киллерах, нормальных плазматических клетках и других иммунных клетках, в том числе на некоторых Т-клетках, моноцитах, В-клетках и дендритных клетках. SLAMF7 не обнаруживается на клетках здоровых тканей и гемопоэтических стволовых клетках.
Элотузумаб напрямую активирует натуральные киллеры через SLAMF7 и Fc-рецепторы и тем самым усиливает их противомиеломную активность in vitro. Элотузумаб также связывается с SLAMF7 на миеломных клетках, что способствует их взаимодействию с натуральными киллерами и уничтожению миеломных клеток посредством антитело-зависимой клеточной цитотоксичности (АЗКЦ). В доклинических исследованиях элотузумаб показал синергетическую активность при совместном применении с леналидомидом.
Отсутствуют данные по канцерогенности или мутагенности препарата у животных и человека.
Фармакокинетика
Фармакокинетику элотузумаба изучали у пациентов с множественной миеломой, получавших препарат в дозах от 0,5 до 20 мг/кг. Элотузумаб обладает нелинейной фармакокинетикой, клиренс препарата снижается с 17,5 до 5,8 мл/день/кг при повышении дозы с 0,5 до 20 мг/кг. Это позволяет предположить, что клиренс зависит от взаимодействия с мишенью, что приводит к более чем пропорциональному увеличению площади под кривой концентрация-время (AUC). Объем распределения элотузумаба приближается к объему внутрисосудистого пространства и не зависит от дозы препарата. По результатам популяционного фармакокинетического анализа типичное значение объёма центрального распределения составляет 4,04 л. При введении элотузумаба в дозе 10 мг/кг, концентрация препарата в крови снижается приблизительно до 3% от ранее достигнутой максимальной равновесной концентрации. На мышиных моделях ксенотрансплантата было установлено, что максимальная терапевтическая эффективность препарата наблюдается при концентрации элотузумаба в плазме крови 70 мкг/мл. У человека применение элотузумаба в дозе 10 мг/кг обеспечивает достижение равновесной концентрации более 70 мкг/мл в плазме крови.
Метаболизм элотузумаба не охарактеризован. Как ожидается, в процессе катаболизма моноклональные антитела будут деградировать на небольшие пептиды и аминокислоты.
Особые группы пациентов
Клиренс элотузумаба возрастает с увеличением массы тела, что является основанием для расчета дозы препарата на массу тела пациента. Популяционный фармакокинетический анализ показал отсутствие зависимости клиренса элотузумаба от возраста (от 37 до 88 лет), пола, расы, исходной активностилактатдегидрогеназы (ЛДГ), концентрации альбумина, нарушения функции почек, а также наличия нарушения функции почек (от легкой степени до тяжелой, с необходимостью гемодиализа или без таковой) или нарушения функции печени легкой степени.
Нарушение функции почек
Фармакокинетические свойства элотузумаба были изучены при назначении комбинированной терапии с ленолидамидом и дексаметазоном у пациентов с множественной миеломой и нормальной функцией почек (клиренс креатинина более 90 мл/мин), тяжелым нарушением функции почек, не требующим проведения диализа (клиренс креатинина менее 30 мл/мин) или терминальной стадией нарушения функции почек, требующей проведения диализа (клиренс креатинина менее 30 мл/мин). Не было отмечено клинически значимых различий в клиренсе препарата у пациентов с тяжелыми нарушениями функции почек (с и без диализа) и пациентами с нормальной функцией почек.
Нарушение функции печени
Фармакокинетические свойства элотузумаба были изучены у пациентов с легкой степенью печеночной недостаточности (общий билирубин менее или равен верхней границе нормы (ВГН) и ACT выше ВГН; или общий билирубин в 1,0-1,5 раза выше ВГН или любое значение ACT) в сравнении с пациентами с нормальной функцией печени (общий билирубин и ACT в пределах нормы). Различий в клиренсе элотузумаба между вышеуказанными группами пациентов не обнаружено. Фармакокинетика препарата не изучалась у пациентов со средней степенью печеночной недостаточности (повышение общего билирубина от 1,5 до 3,0 ВГН, любое значение ACT) и тяжёлой степенью печеночной недостаточности (общий билирубин более 3,0 ВГН, любое значение ACT).
Нарушения ЭКГ
Возможное влияние элотузумаба на удлинения интервала QTc изучали у пациентов при использовании дозировок 10 и 20 мг/кг как в виде монотерапии, так и в комбинации с леналидомидом и дексаметазоном. Изменений средних значений интервала QT обнаружено не было. Анализ клинических показателей ЭКГ и концентрации элотузумаба в сыворотке крови не выявил значимого влияния препарата на реполяризацию. В клинических исследованиях не были отмечены какие-либо клинически значимые изменения частоты сердечных сокращений, длительности интервала PR, длительности комплекса QRS, атрио-вентрикулярной проводимости или деполяризация, а также случаи желудочковой тахикардии типа «пируэт».
ПОКАЗАНИЯ К ПРИМЕНЕНИЮ
Элотузумаб в комбинации с леналидомидом и дексаметазоном показан для лечения пациентов с множественной миеломой, получивших один или несколько предшествующих курсов терапии.
ПРОТИВОПОКАЗАНИЯ
- Гиперчувствительность к любому компоненту препарата.
- Детский возраст до 18 лет в связи с отсутствием данных по эффективности и безопасности.
- Беременность и период грудного вскармливания в связи с отсутствием данных по эффективности и безопасности.
- Нарушение функции печени средней степени и тяжёлой степени тяжести.
- При наличии противопоказаний к применению препаратов комбинированной терапии - см. инструкции по применению соответствующих препаратов.
ПРИМЕНЕНИЕ ПРИ БЕРЕМЕННОСТИ И В ПЕРИОД ГРУДНОГО ВСКАРМЛИВАНИЯ
Применение при беременности
Исследований влияния элотузумаба на репродуктивную функцию у животных не проводилось. Исследований применения Эмплисити® у беременных женщин не проводилось. Применение элотузумаба при беременности противопоказано. Во время лечения женщинам детородного возраста рекомендуется применение контрацепции.
Комбинированная терапия
Применение препарата в комбинации с леналидомидом может вызывать тяжелые, угрожающие жизни врожденные дефекты, поэтому необходимо придерживаться требований, связанных с предохранением от беременности, включая тестирование и контрацепцию. Леналидомид проникает как в кровь, так и в сперму пациентов, поэтому во время лечения как мужчинам, так и женщинам необходимо использовать надежные методы контрацепции. Перед применением комбинированной терапии с леналидамидом необходимо ознакомиться с инструкцией по медицинскому применению препарата леналидомид.
Применение в период грудного вскармливания
Исследований касательно проникновения в грудное молоко женщин в период лактации не проводилось. Согласно общим сведениям попадание антител в грудное молоко возможно, поэтому нельзя исключить риск для новорожденного при применении препарата в период грудного вскармливания. Ввиду потенциальной опасности развития серьезных побочных реакций у ребенка, применение Эмплисити® в период грудного вскармливания противопоказано.
Влияние на фертильность
Исследований касательно влияния элотузумаба на фертильность не проводилось.
СПОСОБ ПРИМЕНЕНИЯ И ДОЗЫ
Препарат должен вводиться под руководством врача, имеющего опыт лечения множественной миеломы.
Перед введением каждой дозы Эмплисти* пациенты должны получать премедикацию.
Режим дозирования
Рекомендуемая доза Эмплисити® в комбинации с дексаметазоном составляет 10 мг/кг в виде внутривенной инфузии в дни 1, 8, 15, 22 в течение первых двух 28-дневных циклов и каждые две недели в последующих циклах в дни 1 и 15. Лечение должно продолжаться до прогрессирования заболевания или до появления признаков непереносимой токсичности. Дексаметазон следует применять следующим образом:
- В дни введения Эмплисити®, дексаметазон следует принимать по 28 мг внутрь за 3-24 часа до введения Эмплисити®, а также 8 мг внутривенно за 45 - 90 минут до введения Эмплисити .
- В дни, когда Эмплисити® не вводят, но назначена доза дексаметазона, его следует принимать в дозе 40 мг внутрь.
- Рекомендуемая доза леналидомида составляет 25 мг внутрь один раз в день в дни 1 -21 каждого 28 дневного цикла, по крайней мере, за 2 часа после введения элотузумаба.
Схема введения доз представлено в Таблице 1.
Таблица 1: Рекомендованная схема введения Эмплисити® в комбинации с леналидомидом и дексаметазоном
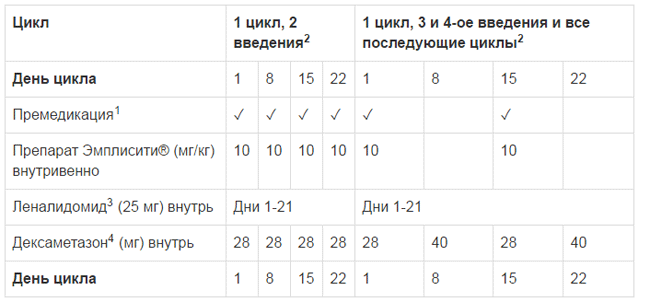
1Премедикация следующими препаратами проводится за 45-90 минут до введения Эмплисити®: 8 мг дексаметазона внутривенно, блокатор Н1-гистаминовых рецепторов: дифенгидрамин (25-50 мг внутрь или внутривенно) или аналогичный препарат; блокатор Н2-гистаминовых рецепторов: ранитидин (50 мг внутривенно или 150 мг внутрь) или аналогичный препарат; парацетамол (650-1000 мг внутрь).
2 Цикл 28 дней
3 Не ранее чем через 2 часа после введения Эмплисити®.
4 Внутрь дексаметазон (28 мг) за 3-24 часа до введения Эмплисити®.
Инструкции по введению препарата
Введение Эмплисити® следует начинать со скоростью 0,5 мл в минуту. При хорошей переносимости скорость ведения можно пошагово увеличивать, как описано в Таблице 2. Максимальная скорость введения не должна превышать 5 мл в минуту.
Таблица 2: Скорость введения Эмплисити®
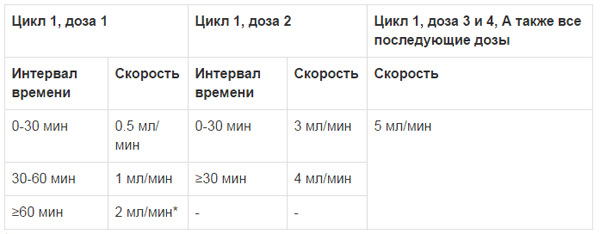
* Поддерживать эту скорость до завершения введения, около 1 часа, в зависимости от массы тела пациента.
Изменение режима введения
Если введение одного из препаратов приостановлено, прервано или прекращено, терапию остальными препаратами можно продолжать по расписанию. Однако, если приостановлено или прекращено применение дексаметазона, то применение Эмплисити® должно быть основано на клинической оценке риска развития реакций гиперчувствительности. При развитии инфузионных реакций 2 степени и выше, введение Эмплисити® необходимо приостановить и назначить соответствующее лечение. После снижения тяжести инфузионной реакции до 1 степени и ниже, введение Эмплисити® следует возобновить со скоростью 0.5 мл в минуту и, при хорошей переносимости, постепенно повышать на 0.5 мл в минуту каждые 30 минут до той скорости, на которой развилась инфузионная реакция. Если повторного развития инфузионных реакций не происходит, можно продолжить увеличение скорости введения по схеме (см. Таблицу 2 Скорость введения Эмплисити®).
У пациентов, у которых ранее развивались инфузионные реакции, показатели жизненно важных функций необходимо контролировать каждые 30 минут в течение 2 часов после окончания введения Эмплисити®. Если инфузионная реакция развивается снова, введение препарата следует прекратить и не возобновлять в этот день. Очень тяжелые инфузионные реакции могут потребовать отмены терапии препаратом Эмплисити и проведения неотложной терапии.
Приостановление терапии и изменение схемы терапии дексаметазоном и леналидомидом следует проводить по клиническим показаниям.
Пациенты с инфузионными реакциями легкой степени тяжести могут получать препарат Эмплисити® со сниженной скоростью введения и под тщательным наблюдением врача.
Правила приготовления раствора
Концентрат и раствор для инфузии готовят в асептических условиях.
Препарат Эмплисити® совместим со следующими типами оборудования для инфузии:
- Стеклянные флаконы, поливинилхлоридные (PVC) и полиолефиновые мешки для инфузии;
- Поливинилхлоридные (PVC) системы для внутривенного введения с автоматическим инфузионным насосом;
- Полиэфирсульфоновые (размер пор: 0,2 - 1,2 мкм) и нейлоновые (размер пор: 0,2 мкм) проточные фильтры для инфузионных систем.
Частично использованные флаконы с препаратом должны быть утилизированы, согласно местным рекомендациям.
Правила приготовления раствора
- Дозировка препарата определяется с учетом веса пациента - 10 мг/кг. На основании дозы, выписанной пациенту, определяют количество флаконов препарата, необходимых для введения. Для приготовления разовой дозы для введения на одного пациента может потребоваться более 1 флакона.
- Удаляют защитную пластиковую крышку с флакона. Пробку флакона протирают стерильной ватой, смоченной в спирте.
- Содержимое каждого флакона растворяют в воде для инъекций как указано в Таблице 3, используя шприц соответствующего объема (размер иглы 18 или меньше).
Таблица 3: Инструкции по разведению растворению Эмплисити®

- Для уменьшения пенообразования воду добавляют медленно, придерживая поршень шприца пальцами. Струю растворителя направляют строго на стенку флакона (а не на лиофилизат).
- Осторожно перемешивают круговыми движениями, держа флакон вертикально. Затем переворачивают флакон, чтобы растворить весь порошок, который может находиться в верхней части флакона или на пробке. НЕ ВСТРЯХИВАТЬ.
- Лиофилизат должен раствориться в течение 10 минут. После полного растворения, оставьте полученный раствор еще на 5-10 минут перед дальнейшим разведением.
- Полученный концентрат представляет собой прозрачную или сильно опалесцирующую жидкость от бесцветного до светло-желтого цвета. Нельзя использовать раствор другого цвета или содержащий инородные частицы. Флаконы с измененной окраской или частицами выбрасывают.
- После растворения отбирают необходимый объем концентрата из каждого флакона для рассчитанной дозы, но не более максимального -16 мл из флакона 400 мг и 12 мл из 300 мг флакона. Дальнейшее разведение проводят во флаконе стерильной инфузионной системы с помощью 230 мл 0,9 % раствора натрия хлорида для инфузий или стерильного 5 % раствора декстрозы для инфузий. Объем 0,9 % раствора натрия хлорида для инфузий или стерильного 5 % раствора декстрозы для инфузий подбирается таким образом, чтобы концентрация элотузумаба в растворе была не более чем 5 мл/кг массы тела пациента при любой вводимой дозе препарата.
- Приготовленный раствор перемешивают путем осторожного переворачивания емкости для инфузий. Не встряхивать флакон перед использованием! Приготовленный раствор нельзя замораживать.
- Перед введением необходимо провести визуальный контроль приготовленного раствора препарата на наличие механических включений и изменение цвета. Раствор Эмплисити® представляет собой прозрачный или сильно опалесцирующий раствор, от бесцветного до светло-желтого цвета.
- Препарат должен вводиться через стерильную инфузионную систему с низкой
- способностью связывания белков со стерильным, апирогенным проточным
- фильтром (размер пор 0,2 - 1,2 мкм) и автоматическим инфузионным насосом.
- Введение Эмплисити® следует начинать со скорости 0,5 мл в минуту. Если введение переносится хорошо, скорость инфузии можно постепенно повышать, как указано в Таблице 2. Максимальная скорость инфузии не должна превышать 5 мл в минуту.
- Препарат нельзя вводить в виде быстрой внутривенной инъекции или в виде болюсной инъекции.
- Не смешивать препарат Эмплисити® с другими лекарственными препаратами в одном флаконе или системе для инфузии и не вводить его одновременно с другими препаратами для инфузии.
- После введения каждой дозы Эмплисити® необходимо промыть инфузионную систему стерильным 0,9 % изотоническим раствором натрия хлорида для инфузии или стерильным 5 % раствором декстрозы для инфузии. С точки зрения микробиологической чистоты, приготовленный раствор должен использоваться немедленно. В противном случае приготовленный раствор можно хранить в защищенном от света месте до 24 часов при температуре 2 - 8 °С (Из указанных 24 часов раствор препарата может находится при комнатной температуре (20-25 °С) и дневном освещении в течение не более чем 8 часов, включая время, необходимое для введения препарата). Приготовленный раствор нельзя замораживать!
- Неиспользованный остаток Эмплисити® во флаконе и пустой флакон необходимо уничтожить.
Пациенты пожилого возраста
В клинических исследованиях различий по безопасности и эффективности между пациентами 65 лет и более и пациентами более молодого возраста (менее 65 лет) отмечено не было. Данные об эффективности и безопасности препарата у больных старше 85 лет ограничены.
Нарушение функции почек
В клинических исследованиях фармакокинетика элотузумаба в комбинированной терапии существенным образом не отличалась у пациентов с нормальной функцией почек, у пациентов с тяжелым нарушением функции почек, которое не требовало проведения диализа, и у пациентов с терминальной стадией нарушения функции почек, которое требовало проведения диализа. У пациентов с нарушениями функции почек разной степени тяжести, а также у пациентов, нуждающихся в проведении диализа, коррекция дозы Эмплитисити® не требуется.
Нарушение функции печени
У пациентов с легкой степенью тяжести печеночной недостаточности коррекция дозы Эмплисити® не требуется. У пациентов с нарушением функции печени средней или тяжелой степени клинические исследования не проводились.
ПОБОЧНОЕ ДЕЙСТВИЕ
Ниже приведены побочные реакции, отмечавшиеся у пациентов с множественной миеломой, получающих комбинированную терапию препаратом Эмплисити8. Большинство нежелательных реакций были легкими или умеренными (1 или 2 степени). Эти реакции представлены по системно-органным классам и по частоте. Частота встречаемости определяется как: очень часто (>1/10); часто (>1/100, <1/10); нечасто (>1/1000, <1/100); редко (>1/10000, <1/1000); очень редко (<1/10000).
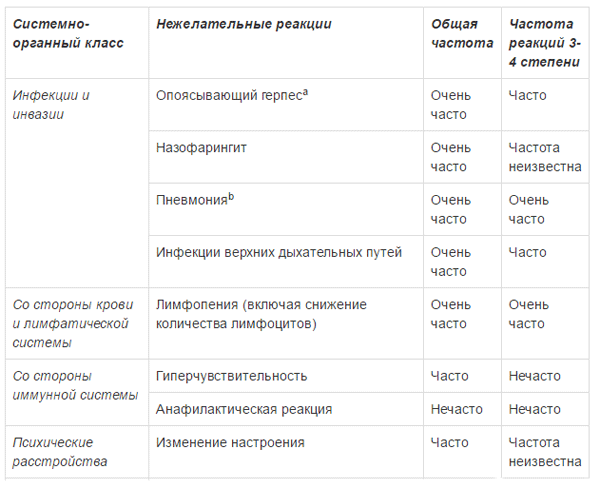
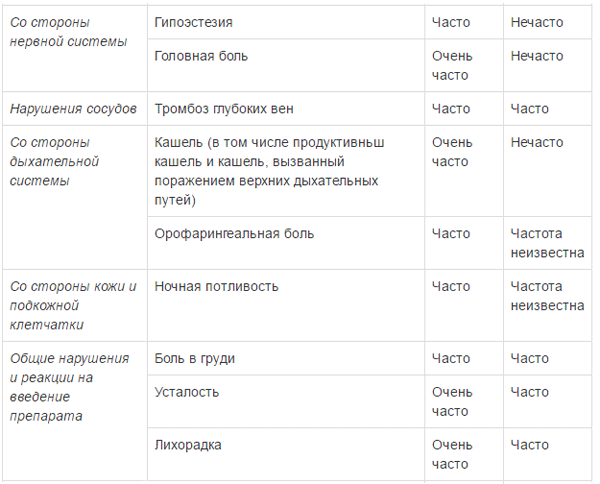

a опоясывающий герпес объединяет группу терминов: герпес Зостера, оральный герпес, герпетические вирусные инфекции b пневмония объединяет группу терминов: пневмония, атипичная пневмония, бронхопневмония, долевая пневмония, бактериальная пневмония, грибковая пневмония, гриппозная пневмония и пневмококковая пневмония
Инфузионные реакции
В клиническом исследовании с участием пациентов с множественной миеломой инфузионные реакции были отмечены примерно у 10% пациентов, которые прошли премедикацию и получавших терапию препаратом Эмплисити в сочетании с леналидомидом и дексаметазоном (N=318). Частота возникновения инфузионных реакций от легкой до средней степени тяжести составила > 50% среди пациентов, которые не прошли премедикацию. Все отчеты по инфузионным реакциям относились к реакции 3 степени и ниже. Инфузионные реакции 3 степени отмечались у 1% пациентов. К наиболее часто отмечаемым симптомам инфузионной реакции относились: жар, озноб, гипертензия. 5% пациентов требовалось прерывание введения Эмплисити в среднем в течение 25 минут ввиду инфузионной реакции, а 1% пациентов прекратил лечение ввиду появления инфузионных реакций. Среди пациентов, у которых была отмечена инфузионная реакция, у 70% (23/33) отмечалось развитие реакции во время получения первой дозы.
Инфекции
Частота возникновения инфекций, включая пневмонию, была выше в группе терапии препаратом Эмплисити®, чем в контрольной группе. В клиническом исследовании с участием пациентов с множественной миеломой инфекции были отмечены у 81,4% пациентов, (К) получавших терапию препаратом Эмплисити в сочетании с леналидомидом и дексаметазоном (N=318), и у 74,4% пациентов, получавших терапию леналидомидом и дексаметазоном (N=317). Инфекции 3-4 степени были отмечены у 28% пациентов, получавших терапию препаратом Эмплисити в сочетании с леналидомидом и дексаметазоном, и у 24,3% пациентов, получавших терапию леналидомидом и дексаметазоном. Смертельные инфекции встречались редко и отмечались у 2,5% пациентов, получавших терапию препаратом Эмплисити® в сочетании с леналидомидом и дексаметазоном, и у 2,2% пациентов, получавших терапию леналидомидом и дексаметазоном. Частота развития пневмонии была выше в группе терапии препаратом Эмплисити в сочетании с леналидомидом и дексаметазоном по сравнению с группой терапии леналидомидом и дексаметазоном: 15,1% и 11,7% пациентов, с летальным исходом 0,6% и 0%, соответственно.
Вторичные злокачественные новообразования
Частота развития вторичных злокачественных новообразований была выше у пациентов, (К) получавших терапию препаратом Эмплисити , чем в контрольной группе. В рамках клинического исследования с участием пациентов с множественной миеломой, инвазивные вторичные злокачественные новообразования отмечались у 6,9% пациентов, получавших терапию препаратом Эмплисити в сочетании с леналидомидом и дексаметазоном (N=318), и у 4,1% пациентов, получавших терапию леналидомидом и дексаметазоном (N=317). Известно, что вторичные злокачественные новообразования связаны с воздействием леналидомида, которое отмечалось в течение более длительного периода времени у пациентов, получавших (К) комбинированную терапию препаратом Эмплисити , леналидомидом и дексаметазоном, чем среди пациентов, получавших терапию леналидомидом и дексаметазоном. Частота возникновения гематологических опухолей была одинаковой в двух группах терапии (1,6%). Солидные опухоли отмечались у 2,5% пациентов, получавших терапию препаратом Эмплисити в сочетании с леналидомидом и дексаметазоном и у 1,9% пациентов, получавших терапию леналидомидом и дексаметазоном. Рак кожи (не меланома) был отмечен у 3,1% пациентов, получавших терапию препаратом Эмплисити в сочетании с леналидомидом и дексаметазоном, и у 1,6% пациентов, получавших терапию леналидомидом и дексаметазоном.
Тромбоз глубоких вен
В клиническом исследовании с участием пациентов с множественной миеломой тромбозы глубоких вен были отмечены у 7,2% пациентов, получавших терапию препаратом Эмплисити в сочетании с леналидомидом и дексаметазоном (N=318), и у 3,8% пациентов, получавших терапию леналидомидом и дексаметазоном (N=317). Среди пациентов, принимавших аспирин, тромбоз глубоких вен был отмечен у 4,1% пациентов, получавших терапию препаратом Эмплисити в сочетании с леналидомидом и дексаметазоном, и у 1,4% пациентов, получавших терапию леналидомидом и дексаметазоном. Частота развития тромбоза глубоких вен, отмеченная среди групп терапии, была сходной для пациентов, получавших профилактическую терапию с применением низкомолекулярного гепарина (2,2% в обеих группах лечения), и для пациентов, получавших антагонисты витамина К, и показатели составили 0% для пациентов, получавших терапию препаратом Эмплисити в сочетании с леналидомидом и дексаметазоном, и 6,7% для пациентов, получавших терапию леналидомидом и дексаметазоном.
Иммуногенность
Препарат Эмплисити*, как и другие белковые препараты, обладает иммуногенностью. У 18,5 % пациентов (получавших терапию препаратом Эмплисити , и у которых измеряли уровень соответствующих антител) наблюдалось образование антител к препарату. Нейтрализующие антитела определялись у 6,4 %. У большинства пациентов иммуногенность препарата была преходящей и наблюдалась только в начале терапии, и исчезала через 2-4 месяца. Не выявлено зависимости между наличием антител к препарату в плазме крови и изменений фармакокинетических параметров, эффективности и безопасности препарата.
ПЕРЕДОЗИРОВКА
Отмечен один случай передозировки препаратом при использовании 23,3 мг/кг элотузумаба в комбинации с леналидомидом и дексаметазоном. При этом у пациента не наблюдалось инфузионных реакций и других симптомов, и не потребовалось какое-либо дополнительное лечение, связанное с передозировкой. Пациент продолжил терапию препаратом Эмплисити®.
Максимально переносимая доза не установлена. В клинических исследованиях примерно 78 пациентов получали элотузумаб в дозе 20 мг/кг без развития токсических эффектов. При передозировке лечение должно заключаться в симптоматической лекарственной терапии в соответствии с возникающими побочными реакциями при тщательном наблюдении за пациентом.
ВЗАИМОДЕЙСТВИЕ С ДРУГИМИ ЛЕКАРСТВЕННЫМИ СРЕДСТВАМИ
Фармакокинетических исследований по изучению лекарственных взаимодействий с препаратом Эмплисити* проведено не было. Препарат представляет собой человеческое моноклональное антитело. Ввиду того, что антитела не подвергаются метаболизму при участии изоферментов цитохрома Р450 и других изоферментов, ингибирование или индукция этих изоферментов не оказывают влияние на фармакокинетику элотузумаба при совместном применении с другими лекарственными препаратами. Однако, ввиду того что препарат Эмплисити® применяется в комбинации с ленолидамидом и дексаметазоном, необходимо ознакомиться с информацией о взаимодействии в инструкциях этих препаратов.
ОСОБЫЕ УКАЗАНИЯ
Инфузионные реакции
Эмплисити® может вызывать развитие инфузионных реакций. Инфузионные реакции были отмечены в клинических исследованиях приблизительно у 10% пациентов, получавших терапию препаратом Эмплисити®, леналидомидом и дексаметазоном.
Все отмеченные инфузионные реакции были не выше 3-ей степени тяжести. В клинических исследованиях при комбинированной терапии с леналидомидом и дексаметазоном инфузионные реакции 3 степени возникли у 1% пациентов. Наиболее частыми побочными реакциями были лихорадка, озноб и гипертензия. В исследовании комбинированной терапии с леналидомидом и дексаметазоном у 5% пациентов потребовалось прерывание введения препарата Эмплисити® из-за развития инфузионных реакций в среднем на 25 минуте, а у 1% пациентов препарат пришлось отменить из-за инфузионных реакций. Среди пациентов, у которых возникли инфузионные реакции, у 70% (в случае комбинации с леналидомидом и дексаметазоном) они возникали во время введения первой дозы.
Комбинированная терапия с использованием препарата
Препарат Эмплисити1 применяется в комбинации с другими препаратами; таким образом,предостережения и меры предосторожности, применяемые к этим препаратам, также будут актуальны и для комбинированной терапии, включающей элотузумаб. В том числе он потенциальный риск для плода, наличие и передачу препарата/антител со спермой и кровью и запрет на донорство крови и/или спермы. Перед началом лечения необходимо учесть указания по применению всех препаратов, применяемых в комбинации с препаратом Эмплисити®.
Инфекции
В клинических исследованиях с участием пациентов с множественной миеломой частота возникновения инфекций, включая пневмонию, была выше среди пациентов, получавших терапию препаратом Эмплисити®. Необходимо следить за состоянием пациентов и применять терапию для лечения инфекционных заболеваний.
Вторичные злокачественные новообразования
В клиническом исследовании с участием пациентов с множественной миеломой, в котором проводилось сравнение терапии препаратом Эмплисити" в сочетании с леналидомидом и дексаметазоном и терапии леналидомидом и дексаметазоном, частота появления вторичных злокачественных новообразований, а в частности солидных опухолей и рака кожи (не меланома), была выше среди пациентов, проходивших терапию препаратом Эмплисити . Известно, что вторичные злокачественные новообразования связаны с воздействием леналидомида, которое было в течение более длительного периода времени у пациентов, проходивших терапию препаратом Эмплисити в сочетании с леналидомидом и дексаметазоном, чем среди пациентов, получавших леналидомид и дексаметазон. Частота возникновения гематологических опухолей была одинаковой в двух группах терапии. Необходимо контролировать пациентов на предмет развития вторичных злокачественных новообразований.
ВЛИЯНИЕ НА СПОСОБНОСТЬ УПРАВЛЯТЬ ТРАНСПОРТНЫМИ СРЕДСТВАМИ, МЕХАНИЗМАМИ
Исследований влияния применения препарата на способность управлять транспортными средствами и работать с механизмами не проводилось.
ФОРМА ВЫПУСКА
Лиофилизат для приготовления концентрата для приготовления раствора для инфузий 300 мг, 400 мг.
По 340 мг или 440 мг активного вещества во флакон прозрачного бесцветного стекла типа I, укупоренный бутилрезиновой пробкой и алюминиевым колпачком с защитной пластиковой крышкой.
По 1 флакону вместе с инструкцией по применению помещают в пачку картонную. Места вскрытия картонной пачки заклеены двумя защитными прозрачными бесцветными стикерами; на среднюю часть каждого стикера, ограниченную линиями отрыва, нанесен рисунок в виде желтой сетки.
УСЛОВИЯ ХРАНЕНИЯ
Хранить при температуре от 2 до 8 °С в защищенном от света месте.
Не замораживать.
Хранить в недоступном для детей месте!
СРОК ГОДНОСТИ
3 года.
Не применять по истечении срока годности.
УСЛОВИЯ ОТПУСКА
Отпускают по рецепту.
ЮРИДИЧЕСКОЕ ЛИЦО, НА ИМЯ КОТОРОГО ВЫДАНО РЕГИСТРАЦИОННОЕ УДОСТОВЕРЕНИЕ
Бристол-Майерс Сквибб Компани, США
345, Парк-авеню, г. Нью-Йорк, штат Нью-Йорк, США
Bristol-Myers Squibb Company, USA
345, Park Avenue, New York, New York, USA
ПРОИЗВОДИТЕЛЬ/ ФАСОВЩИК (ПЕРВИЧНАЯ УПАКОВКА)
Бристол-Майерс Сквибб Холдинге Фарма, Лтд. Лиабилити Компани
Роуд 686 Км 2,3 Бо. Тиеррас Нуевас, Манати, Пуэрто-Рико, 00674, США
Bristol-Myers Squibb Holdings Pharma, Ltd. Liability Company,
Road 686 Km 2.3 Bo. Tierras Nuevas, Manati, Puerto Rico, 00674, USA
УПАКОВЩИК (ВТОРИЧНАЯ УПАКОВКА)/ВЫПУСКАЮЩИЙ КОНТРОЛЬ КАЧЕСТВА
Бристол-Майерс Сквибб С.р.Л., Италия
Локалита Фонтана дель Черазо, 03012, Ананьи (ФР), Италия
Bristol-Myers Squibb S.r.L., Italy
Localita Fontana del Ceraso, 03012, Anagni (FR), Italy
Претензии потребителей направлять по адресу Представительства в РФ:
ООО «Бристол-Майерс Сквибб», 105064, г. Москва, Земляной вал, д.9
Комментарии
ПРАКТИКА ПЕДИАТРА



