Плейотропный нейропротективный и метаболический эффекты Актовегина
СтатьиFausto Machicao1, Dafin Fior Muresanu2, Harald Hundsberger3, Maren Pfluger3, Alla Guekht4
1 Molecular Genetics and Diagnosis, Department of Internal Medicine IV, Otfried Muller Str. 10, University Hospital, D-72076, Тюбинген, Германия;
2 Department of Neurosciences, University of Medicine and Pharmacy «Iuliu Hatieganu», Клуж-Напока, Румыния;
3 Medical and Pharmaceutical Biotechnology IMC Krems, University of Applied Sciences Krems, Piaristengasse 1, A-3500, Кремс, Австрия;
4 Neurology and Rehabilitation, Department of Neurology and Neurosurgery of the Russian State, Москва, Россия
Перевод: С.С. Никитин . Опубликован в журнале "Нервно-мышечные болезни", №4, 2012
В обзоре рассматриваются механизмы действия Актовегина в контексте изучения его эффектов на доклиническом уровне и новой концепции фармакологического лечения неврологических расстройств. Актовегин, получаемый при ультрафильтрации крови телят, состоит из более чем 200 биологических субстанций. Препарат используется при широком спектре заболеваний, включая нарушения периферического и мозгового кровообращения, ожоги, плохое заживление ран, радиационные поражения и диабетическую полинейропатию. Актовегин состоит из молекул малого размера, которые находятся в организме в нормальных физиологических условиях, и поэтому исследования их фармакокинетики и фармакодинамики для определения активной субстанции препарата затруднены. Результаты преклинических исследований показали, что Актовегин улучшает метаболический баланс путем повышения усвоения глюкозы и потребление кислорода в условиях ишемии. Актовегин также повышает устойчивость к гамма-радиации и стимулирует ранозаживление. В более поздних работах было установлено, что антиоксидантный и антиапоптотический механизмы действия лежат в основе нейропротективных свойств Актовегина, подтвержденных в экспериментах на первичных нейронах гиппокампа крыс и стрептозотоцининдуцированной модели диабетической полинейропатии у крыс. Последние данные свидетельствуют о положительном влиянии Актовегина на фактор NF-kB, но при этом многие молекулярные и клеточные механизмы его действия остаются неизвестными. В первую очередь это касается влияния Актовегина на нейропластичность, нейрогенез и трофическую функцию нервной системы, и данный аспект требует дальнейших исследований. Тем не менее становится очевидным, что мультифакториальная и многокомпонентная природа Актовегина определяет его плейотропный нейропротективный механизм действия и клиническую эффективность.
Ключевые слова: Актовегин, метаболический эффект, механизм действия, нейропротекция, апоптоз, оксидативный стресс
1. Введение
До сих пор вопрос об оптимальных терапевтических подходах защиты мозга и восстановления его функциональных нарушений остается открытым, особенно если учесть, что до конца остаются неясными все детали эндогенных процессов и патофизиологических механизмов, участвующих в работе поврежденного мозга. В связи с этим мы по-прежнему используем упрощенную модель при рассмотрении наблюдаемых нарушений.
Так называемый редукционистский подход в понимании формирования мозговых нарушений исходит из того, что наблюдаемые изменения являются результатом независимого линейного развития таких процессов, как эксайтотоксичность, воспаление, апоптоз и оксидативный стресс [1].
Классическая концепция нейропротекции подразумевает подавление отдельного патофизиологического механизма при использовании соответствующего препарата. Данная терапевтическая стратегия основана на том, что если отдельно взятый патофизиологический механизм может быть подавлен фармакологическим агентом, то в этом случае вызываемые этим процессом нарушения будут исключены из общего объема нарушений, вызываемых всеми возможными патофизиологическими механизмами. Однако, как следует из клинических исследований по нейропротекции, в случаях с химическими препаратами данная субстракционная стратегия не работает [2, 3].
Тенденция ограничения эффекта лекарственного препарата рамками только одного механизма действия и одним фокусным эффектом может привести к игнорированию других, не менее эффективных свойств, а также затруднить разработку более эффективных терапевтических подходов. При рассмотрении фармакологических подходов защиты и восстановления работы мозга необходима смена концепции. Сегодня нейротрофичность, нейропротекция, нейропластичность и нейрогенез рассматриваются как фундаментальные нейробиологические процессы, участвующие в реализации эндогенной защитной активности (ЭЗА), а также в попытках противодействовать патофизиологическим повреждающим механизмам (ППМ) и стимулировании эндогенного восстановления (рис. 1) [4].
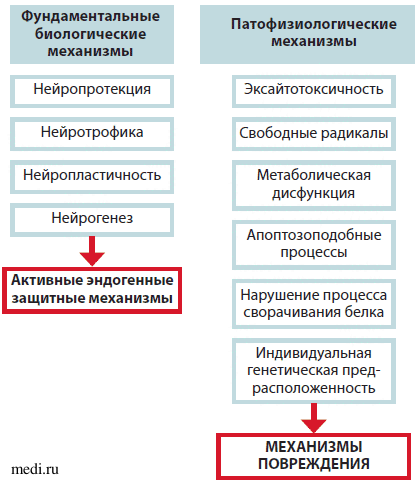 Рис. 1. Эндогенные защитные и повреждающие механизмы
Рис. 1. Эндогенные защитные и повреждающие механизмы
Новая теория защиты и восстановления нарушенной функции мозга основана на том, что с нейробиологических позиций процессы ЭЗА и ППМ имеют общие биологические механизмы реализации. Например, в реализации эксайтотоксичности (патофизиологический механизм) и нейропластичности (нейрорепаративный процесс) участвуют NMDA-рецепторы. Так, в случае фармакологического подавления одного из патофизиологических процессов (например, эксайтотоксичности или воспаления) химическим препаратом, действующим только на 1 звено, одновременно на долгое время нарушаются эндогенные механизмы восстановления (например, нейропластичность и нейротрофика) [1].
Поэтому не рекомендуется рассматривать отдельно фармакологическую нейропротекцию и восстановление нервной ткани, поскольку подобная дихотомия приводит к ошибкам в основных клинических исследованиях. В клинической практике в конечном итоге оценивается общий эффект суммарного действия как протективных, так и восстановительных механизмов ЭЗА, которые на самом деле неразделимы [1].
Предлагаемые соображения включают интегративный фармакологический подход, сфокусированный на веществах с мультимодальной активностью и плейотропными нейропротективными свойствами, на биологических препаратах, обладающих больше чем одним механизмом действия в отличие от химических искусственно созданных малых молекул [5].
Актовегин — депротеинизированный ультрафильтрат, получаемый из крови телят, не содержащий эндотоксинов и антигенов и состоящий из более 200 биологически активных компонентов. Сложный и длительный процесс производства Актовегина включает 2 этапа ультрафильтрации, в которых используются фильтры для выделения молекул разного размера. Сначала отсеиваются молекулы весом 5000 Да с последующим вакуумным удалением оставшегося преципитата с помощью фильтра (0,45 мкм) и титрованием при pH 6,4. Затем, после предварительной фильтрации фильтрами 0,2 и 0,45 мкм проводят стерильную фильтрацию до полной депротеинизации продукта, который в конечном итоге проверяется на отсутствие протеина с использованием электрофореза на SDS-полиакриамидном геле. Молекулярный вес конечного отфильтрованного продукта не превышает 5000 Да.
Состав Актовегина был проверен с использованием самых современных аналитических методик, включая газовую хроматографию и высокоэффективную жидкостную хроматографию в сочетании с масс-спектрометрией. Данные количественных методов анализа возможных метаболитов (Biocrates Life Sciences AG, Инсбрук, Австрия) показали, что Актовегин является комбинацией из более чем 200 биоактивных молекул. Актовегин в основном состоит из субстанций с низким молекулярным весом, включая аминокислоты, биогенные амины и полиамины, сфинголипиды, гексозы, эйкозаноиды, лактат, сукцинат, холин, витамины, аденозинмонофосфат и инозитолфосфоолигосахариды. Также обнаруживаются небольшие количества ацилкарнитинов, фосфолипидов, свободных жирных кислот и оксистеролов; в еще меньшей концентрации представлены простагландины, окисленные полине-насыщенные жирные кислоты и желчные кислоты (неопубликованные данные).
Если учитывать то, что Актовегин по сути является биологическим препаратом, состоящим из молекул, которые содержатся в физиологических условиях, фармакокинетические и фармакодинамические исследования, направленные на определение активных молекул, не представляются возможными. Несмотря на это, экспериментальные данные подтверждают, что Актовегин как препарат имеет множество активных составляющих, которые участвуют во многих внутриклеточных процессах и влияют на специфические пути метаболизма клетки. Однако связь между множественными молекулярными механизмами действия и оказываемыми положительными эффектами in vitro и in vivo изучена лишь частично.
2. Эффекты Актовегина in vitro и in vivo
В ходе ранних доклинических исследований было показано, что Актовегин влияет на процессы заживления ран путем стимуляции роста клеток, включая синтез коллагена и уменьшение дезинтегративных процессов в матриксе [6, 7]. Актовегин также оказывает положительное регенеративное действие при лечении радиационных повреждений [8], а также при нарушениях кровообращения [9, 10]. Однако ней-ропротективные свойства препарата, связанные с отдельными патофизиологическими клеточными механизмами, были рассмотрены лишь недавно.
Было показано, что Актовегин обладает нейропротективным действием, что в культуре нейронов проявляется в увеличении числа живых нейронов и синаптических контактов. В исследовании in vitro [11], нейроны гиппокампа были выделены из мозга крысы и далее культивированы в оптимальных условиях в присутствии разных концентраций Актовегина (0,3—1000 мкг/мл). Обычно культивируемые нейроны гиппокампа быстро погибают в результате апоптоза. Однако через 10 дней в культуре нейронов после применения Актовегина в концентрации больше 10 мкг/мл было отмечено дозозависимое увеличение числа живых клеток в 2,4 раза по сравнению с контрольной культурой. Параллельно с этим методом экспрессии маркерного протеина VGlut1 в пресинаптических терминалях оценивали число возбуждающих синапсов между нейронами. Методами иммуногистохимического анализа выявлено повышение содержания VGlut1 до 3,6 раза на 1 клетку при дозах Актовегина больше 300 мкг/мл, которое также носило дозозависимый характер.
Нейропротективный эффект Актовегина также продемонстрирован в периферической нервной системе при тяжелой нейропатии на модели стрептозотоцининдуцированного (СТЗ-индуцированного) диабета у крыс [12]. Лечение диабетической нейропатии у крыс при интраперитонеальном введении Актовегина в дозах, эквивалентных дозам, используемым в клинических исследованиях у человека, приводило к существенному снижению скорости дегенерации периферических нейронов. Положительный эффект препарата выявлен при гистоморфологическом исследовании кожи лапы крысы в виде увеличения на 32 % плотности распределения внутриэпидермальных нервных волокон по окончании курса терапии Актовегином. Более того, значительное улучшение функционального состояния сенсорных нервных волокон периферических нервов показано при исследовании скорости проведения импульса по сенсорным волокнам в 91 % случаев у крыс, получавших Актовегин. N-ацетилцистин был использован в качестве положительного контроля и, что интересно, оказался бесполезным в данном случае.
3. Влияние Актовегина на усвоение глюкозы и аэробный метаболизм в головном мозге
Доклинические исследования показали, что на молекулярном уровне Актовегин улучшает утилизацию и захват кислорода, также как и энергетический метаболизм и усвоение глюкозы [13—15]. Глюкоза служит основным источником энергии для мозга и для центральной нервной системы (ЦНС); переносчики глюкозы играют важную роль в нейрональном гомеостазе и утилизации глюкозы [16—18]. Нарушение гомеостаза глюкозы в ЦНС приводит к нарушению нейрональной активности и когнитивных функций, что особенно опасно для гиппокампа, области мозга, связанной с обучением и памятью. Рецепторы инсулина и инсулинчувствительные переносчики глюкозы, такие как GLUT4, являются ключевыми составляющими утилизации глюкозы в ЦНС. Они экспрессированы в таких областях мозга, как гиппокамп и мозжечок [19], а также в эндотелии гематоэнцефалического барьера (ГЭБ) [20]. Фундаментальные и клинические исследования показали четкую связь между уровнем инсулина в мозге и когнитивными функциями, включая обучение и память [21]. Например, нарушение трансдукции нейрональных рецепторов инсулина показано при болезни Альцгеймера. S. Hoyer и соавт. показали, что возникающие тяжелые нарушения метаболизма кислорода при внутрицеребровентрикулярном введении в мозг диабетогенного агента приводят к поведенческим нарушениям [22—24]. Это согласуется с гипотезой Salkovic-Petrisic и Hoyer [25] о том, что инсулинрезистентное состояние в мозге может приводить к клеточным и молекулярным нарушениям, наблюдаемым при болезни Альцгеймера.
Актовегин положительно влияет на утилизацию глюкозы и кислорода, что приводит к усилению энергетического метаболизма головного мозга. Показано, что инозитолфосфоолигосахариды могут играть важную роль в регуляции инсулинзависимых энзимов [26, 27]. В эксперименте Актовегин повышал усвоение и утилизацию кислорода [28], что может приводить к стабилизации клеток ЦНС в условиях ишемии и снижать образование лактата. Он также положительно влияет на такие переносчики глюкозы, как GLUT1 и GLUT4, которые при цереброваскулярных нарушениях способствуют улучшению транспорта глюкозы через ГЭБ.
Париетальная кора мозга ответственна за обработку зрительной информации и пространственно-ориентированного внимания, и сокращение площади данной области приводит к деменции. В клинических исследованиях показана способность Актовегина улучшать когнитивные процессы в париетальной коре при возрастных нарушениях памяти, возможно, в результате уменьшения гипоксии мозга [29]. В подтверждение этих клинических наблюдений на модели церебральной ишемии Hoyer и Betz [30] показали, что Актовегин обеспечивает защиту клеток от гипоксии.
4. Снижение апоптоза, обусловленного пептидом Aβ25—35
Амилоидные β-пептиды (Аβ) — основные компоненты амилоидных бляшек, являющихся маркерами болезни Альцгеймера. Нейротоксический пептид повышает оксидативный стресс и активирует воспаление, что в конечном итоге приводит к гибели клетки [31]. Однако Aβ-фрагменты существенно различаются по своим патогенным свойствам. Например, фрагменты Aβ25—35 высокотоксичны [32, 33] и обладают более быстрыми агрегационными способностями по сравнению со всеми остальными изученными амилоидными пептидами [34]. Это было показано в исследованиях на моделях животных, у которых пептиды Aβ25—35 нарушали кратковременную память и приводили к нейропатологическим изменениям, заканчивающимся гибелью нейронов [35, 36]. Вероятно, что цитотоксические и патологические свойства Aβ25—35 лежат в основе нейродегенеративных изменений при болезни Альцгеймера и других нарушениях, но это предположение требует дальнейшего подтверждения.
В недавнем исследовании потенциального механизма действия Актовегина были использованы пептиды Aβ25—35 для определения антиапоптотического эффекта препарата на примере нейрональных клеток in vitro [11]. Первичные нейроны гиппокампа крысы культивировались с Aβ25—35 в присутствии Актовегина с целью определения активности каспазы-3, отражающей степень нейронального апоптоза. В отсутствие Aβ25—35уровень апоптоза не отличался в клетках без применения Актовегина и с его применением. В нейронах гиппокампа, подверженных воздействию Aβ25—35и обработанных возрастающими дозами Актовегина, уровень активированной каспазы-3 достоверно снижался при концентрациях Актовегина менее 300 мкг/ мл (p < 0,001), и наблюдаемое снижение имело дозозависимый характер [11].
Имеются факты, поддерживающие гипотезу о том, что Aβ25—35играют ключевую роль в патогенезе болезни Альцгеймера [37], поэтому угнетение апоптоза, вызванного этими пептидами, расширяет наши представления о механизмах нейропротекции Актовегина.
5. Влияние Актовегина на процессы активации фактора NF-kB
Транскрипция фактора NF-kB играет важную роль в деятельности центральной и периферической нервной системы млекопитающих. Роль фактора NF-kB особенно важна в регуляции воспалительных процессов, которые усугубляют такие состояния, как ишемия и болезнь Альцгеймера, а также в развитии боли, в процессах обучения и памяти и, что особенно важно, в нейропротекции [38—41]. Регуляция активности фактора NF-kB осложняется тем, что нарушения метаболизма могут повышать или понижать его активность и, соответственно, давать различные эффекты в разных тканях.
Участие NF-kB в механизмах нейропротекции опосредуется провоспалительными цитокинами [42]. Показано, что воздействие на первичные нейроны гиппокампа фактором некроза опухоли а (ФНО-а) предотвращает гипоксию и индуцированную оксидом азота нейрональную клеточную гибель посредством увеличения NF-кВ-зависимой экспрессии антиапоптотических факторов Bcl-2 и Bcl-x [43]. В то же время было показано, что ФНО-а также может вызывать нейрональный апоптоз и запускать процесс дегенерации [44], тогда как растворимый ФНО-рецептор-1 может вызывать апоптоз моноцитов [45]. In vivo показано, что предварительное введение ФНО-а крысам с фокальной ишемией снижает степень повреждения мозга [46], в то время как введение трансгенного рецептора ФНО у нокаутных мышей вызывает обширные инфаркты [47]. Данное наблюдение может быть объяснено существованием рецепторов ФНО 2 типов: R1 — вызывающие демиелинизацию и увеличивающие гибель клеток и R2 — вызывающие ремиелинизацию и участвующие в нейропротекции путем активации олигодендроцитов и выделения нейротрофического фактора мозга и цилиарного нейротрофического фактора [5, 48].
В ходе другого исследования, подтверждающего нейропротективные свойства NF-kB, предположили, что данный фактор защищает клетку от гибели в присутствии пептидов Ар и, таким образом, влияет на механизмы апоптоза [40, 49]. На модели грызунов также показано, что снижение экспрессии ингибиторной каппа бета, центрального компонента NF-kB сигнального пути, приводит к потери нейропротекции и может нарушать процессы обучения и памяти [50].
Hundsberger и Pfluger (FH Krems, Австрия) провели новое исследование in vitro для выяснения потенциальной способности Актовегина модулировать активность NF-kB. В качестве экспериментальной системы была использована хорошо известная линия эмбриональных клеток почек человека CellSensor® и NF-KB-bla HEK 293T, которая содержит стабильный трансформированный ген-репортер β-лактамазы, контролируемый элементами NF-kB. Для исключения побочных осмотических явлений Актовегина на клетки в качестве плацебо использован солевой раствор в концентрации эквимолярной раствору Актовегина. Методом флюоресцентной эмиссии показано, что Актовегин активирует ген-репортер экспрессии NF-kB, причем в дозозависимой степени (табл. 1; неопубликованные данные). Эффективная концентрация Актовегина соответствует концентрации ФНО-а приблизительно 400 пг/мл, обладающей тем же эффектом. Полученные данные совпадают с результатами исследования Elmlinger и соавт. [11], в связи с чем предполагают, что нейропротективные и антиапоптотические свойства Актовегина хотя бы частично могут быть связаны с транзиторной активацией NF-kB.
Таблица 1.
Ответ гена-репортера β-лактамазы (в % от исходного уровня нестимулированных клеток) в гене-репортере NF-KB-bla клеток HEK293T на введение Актовегина, стимуляцию ФНО-а и плацебо. Результаты представлены как средние значения от 5 лунок на микропланшете; эксперимент повторяли 3 раза (Hundsberger and Pfluger)
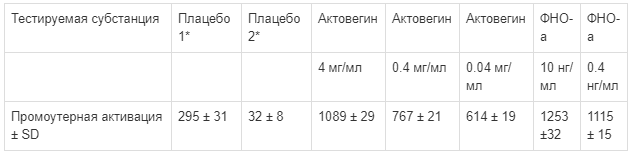 * Осмомолярность плацебо 1 и 2 соответствовала дозировке Актовегина 0,4 мг/мл.
* Осмомолярность плацебо 1 и 2 соответствовала дозировке Актовегина 0,4 мг/мл.Оксидантный стресс вызывает поломку 1 нити ДНК, что приводит к активации ядерного фермента полимеразы поли-АДФ-рибозы (ПАРП). ПАРП играет ключевую роль в обнаружении и репарации повреждений ДНК, но при этом показано, что избыточная активация ПАРП имеет негативные последствия в виде запуска последовательных клеточных процессов, которые в конечном итоге останавливают гликолиз и процесс митохондриального дыхания, что приводит к гибели клетки вследствие энергетического истощения [51, 52].
В эксперименте показано, что сбой генного кодирования ПАРП защищает грызунов от ряда патофизиологических процессов [52—54]. Показано, что нарушение в гене ПАРП обеспечивает защиту нейронов от фокальной ишемии [52] и предотвращает прогрессирование и развитие экспериментального СТЗ-инду-цированного диабета [53, 54]. Дальнейшие исследования подтвердили роль метаболизма ПАРП как важного механизма в развитии дисфункции эндотелия при диабете [55], и недавно было показано, что ПАРП может участвовать и в развитии диабетической полинейропатии. Ингибирование ПАРП у крыс уменьшает нарушение проведения импульса по периферическим нервам [56]. Обобщая эти данные, можно сделать предположение о важной фундаментальной роли ПАРП при таких состояниях, как цереброваскулярные заболевания и диабетическая полинейропатия.
Elmlinger и соавт. в своем исследовании показали положительное влияние Актовегина на оксидантный стресс в первичных нейронах гиппокампа, методом флюоресцентного анализа (по изменению общего содержания активных форм кислорода — АФК). В нейронах, обработанных возрастающими концентрациями третбутилгидропероксида (> 0,2 мМ), обнаружено повышение уровней внутриклеточных АФК (р < 0,001), но в случае использования Актовегина в культуре нейронов отмечено дозозависимое снижение выраженности оксидантного стресса через 10 дней (р < 0,001 при концентрациях > 0,3 мкг/мл) [11].
В исследованиях in vitro влияние Актовегина на анализируемые параметры при экспериментальной диабетической полинейропатии соответствовали результатам, полученным в данном исследовании. У крыс с СТЗ-индуцированным диабетом, которые получали Актовегин интраперитонеально с 11-го по 40-й день после введения стрептозотоцинина, при введении Актовегина в дозе 600 мг/кг отмечалось достоверное снижение активности ПАРП, определяемой по ее содержанию [12]. Как уже упоминалось выше, снижение активности ПАРП при введении Актовегина может лежать в основе улучшения функциональных и морфологических параметров периферической и центральной нервной системы. Результаты, полученные в 2 обсуждаемых выше исследованиях, также говорят о том, что антиоксидантные свойства Актовегина, возможно, и определяют его эффекты в отношении активации ПАРП, что, однако, требует дальнейшего подтверждения в исследованиях.
7. Обсуждение
В представленном обзоре показано, каким образом Актовегин улучшает баланс клеточного метаболизма и влияет на целый ряд патофизиологических механизмов, участвующих в развитии различных нарушений. Поскольку развитие неврологических нарушений определяется целой совокупностью патофизиологических событий, для их устранения необходим интегрированный фармакологический подход, а не упрощенное однонаправленное воздействие. В качестве биологического агента, обладающего плейотропными нейропротективными и метаболическими эффектами (табл. 2) Актовегин со своими механизмами действия соответствует концепции об интегративном терапевтическом подходе.
Таблица 2.
Обобщенные результаты изучения нейропротективного действия Актовегина на доклиническом уровне
Эффект Актовегина | Исследование |
Ускорение ранозаживления | Mochida et al. [6]; Schonwald et al. [7] |
Улучшение кровообращения | Hegner [9]; Giarola [10] |
Протекция от радиационного повреждения | Basu et al. [8] |
Улучшение утилизации кислорода | Reichel et al. [28]; de Groot et al. [29]; Hoyer and Betz [30] |
Улучшение метаболизма глюкозы | Machicao et al. [26] |
Увеличение интрадермальной плотности распределения нервных волокон | Dieckmann et al. [12] |
Увеличение числа выживших нейронов | Elmlinger et al. [11] |
Снижение оксидантного стресса | Elmlinger et al. [11]; Dieckmann et al. [12] |
Снижение апоптоза | Elmlinger et al. [11] |
Улучшение скорости проведения по периферическим нервам | Dieckmann et al. [12] |
6. Оксидантный стресс и метаболизм полимеразы поли-АДФ-рибозы
Плейотропный нейропротективный эффект предполагает одновременное модулирующее влияние на разные повреждающие патологические механизмы (эксайтотоксичность, воспаление, апоптоз, оксидантный стресс и многие другие). В противоположность этому молекулы, обладающие односторонним действием, способны влиять только на какой-либо один патофизиологический механизм. Плейотропный протективный и метаболический эффекты Актовегина кратко освещены в настоящем обзоре. Актовегин оказывает нейропротективное действие на нейроваскулярный функциональный блок за счет своего анти-апоптотического и антиоксидантного действия. Также показан положительный эффект Актовегина на утилизацию глюкозы и кислорода, который приводит к повышению метаболизма в головном мозге. Схематично плейотропный нейропротективный эффект Актовегина представлен на рис. 2.
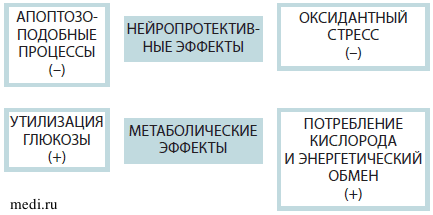 Рис. 2. Плейотропный нейропротективный и метаболические эффекты Актовегина
Рис. 2. Плейотропный нейропротективный и метаболические эффекты Актовегина
Прочие эффекты на другие эндогенные нейробиологические процессы, такие как нейротрофика, нейропластичность и нейрогенез, еще предстоит изучить в будущих исследованиях.
8. Заключение
Актовегин состоит из более чем 200 биологических субстанций, которые влияют на многие внутриклеточные процессы и пути метаболизма. Многочисленные исследования позволили раскрыть возможные механизмы, лежащие в основе нейропротективного и метаболического действия препарата; продемонстрирована способность Актовегина модулировать активацию NF-kB, и тем самым уменьшать апоптоз, подавлять активность ПАРП и положительно влиять на утилизацию как глюкозы, так и кислорода. Однако влияние Актовегина на другие метаболические процессы, потенциально участвующие в нейротрофике, нейропластичности и нейрогенезе, остаются предметом будущих исследований. Тем не менее комплексный состав препарата, имеющего биологическую природу, определяет его плейотропные нейропротективные и метаболические свойства.
Конфликт интересов
Все авторы получили гонорар от фирм Nycomed и Takeda за участие в проведении научных клинических исследований, чтение лекций и консультации.
Благодарность
Авторы выражают искреннюю благодарность Martin Elmlinger, Wolfgang Schonhofer и Gunter Dallinger из Takeda Pharmaceuticals и Alexander Kroll из Synergy Vision за их научный вклад и помощь в редактировании на этапе подготовки рукописи к печати при поддержке Takeda Pharmaceuticals International GmbH, Цюрих, Швейцария.
ЛИТЕРАТУРА
1. Muresanu DF. Neuromodulation with pleiotropic and multimodal drugs - future approaches to treatment of neurological disorders. Acta Neurochir Suppl 2010;106:291-4.
2. Labiche LA, Grotta JC. Clinical trials for cytoprotection in stroke. NeuroRx Jan. 2004;1(1):46-70.
3. Maas AI, Roozenbeek B, Manley GT. Clinical trials in traumatic brain injury: past experience and current developments. Neurotherapeutics 2010 Jan;7(1):115-26.
4. Bornstein NM. Stroke: practical guide for physicians. S. Karger AG; 2009.
5. Muresanu DF. Neuroprotection and neuroplasticity - a holistic approach and future perspectives. J Neurol Sci 2007 Jun 15;257(1-2):38-43.
6. Mochida H, Kikuchi T, Tanaka H, Ikeda A, Fujii Y, Sasamura T, et al. Influence of Actovegin containing infusion solutions on wound healing and function of the intestinal tract in rats. Pharmacol Ther 1989;17:789-97.
7. Schonwald D, Sixt B, Machicao F, Marx E, Haedenkamp G, Bertsch S. Enhanced proliferation of coronary endothelial cells in response to growth factors is synergized by hemodialysate compounds in vitro. Res Exp Med (Berl) 1991;191(4):259-72.
8. Basu SK, Srinivasan MN, Chuttani K, Ghose A. Evaluation of some radioprotectors by the survival study of rats exposed to lethal dose of whole body gamma radiation. J Radiat Res (Tokyo) 1985 Dec;26(4): 395-403.
9. Hegner N. Wirkung eines deproteinisierten Hamoderivates (Actovegin) im induzierten hypovolamischen Schock unter besonderen Berucksichtigung des Energiestoffwechsels. Johannes-Gutenberg Universitat Mainz: Institut fur Anasthesiologie; 1983.
10. Giarola P. Effects of blood extract on plasma lipids, blood coagulation, fibrinolysis, and platelet aggregation in experimental hypercholesterolemia of rabbits. Arzneim-Forsch 1974;24:925-8.
11. Elmlinger MW, Kriebel M, Ziegler D. Neuroprotective and anti-oxidative effects of the hemodialysate actovegin on primary rat neurons in vitro. Neuromolecular Med 2011 Dec;13(4):266-74.
12. Dieckmann A, Kriebel M, Andriambeloson E, Ziegler D, Elmlinger M. Treatment with Actovegin® improves sensory nerve function and pathology in streptozotocin- diabetic rats via mechanisms involving inhibition of PARP activation. Exp Clin Endocrinol Diabetes 2011 Oct 21;120(3):132-8.
13. Kuninaka T, Senga Y, Senga H, Weiner M. Nature of enhanced mitochondrial oxidative metabolism by a calf blood extract. J Cell Physiol 1991 Jan;146(1):148-55.
14. Buchmayer F, Pleiner J, Elmlinger MW, Lauer G, Nell G, Sitte HH. Actovegin®: a biological drug for more than 5 decades. Wien Med Wochenschr 2011 Feb; 161(3-4):80-8.
15. Bachmann W, Forster H, Mehnert H. Experimental studies in animals on the effect of a protein-free blood extract on the metabolism of glucose. Arzneim-Forsch 1968;18:1023-7.
16. Rao J, Oz G, Seaquist ER. Regulation of cerebral glucose metabolism. Minerva Endocrinol 2006 Jun;31(2):149-58.
17. McEwen BS, Reagan LP. Glucose transporter expression in the central nervous system: relationship to synaptic function. Eur J Pharmacol 2004 Apr 19;490(1-3): 13-24.
18. Grillo CA, Piroli GG, Hendry RM, Reagan LP. Insulin-stimulated translocation of GLUT4 to the plasma membrane in rat hippocampus is PI3-kinase dependent. Brain Res 2009 Nov 3;1296:35-45.
19. Kobayashi M, Nikami H, Morimatsu M, Saito M. Expression and localization of insulin-regulatable glucose transporter (GLUT4) in rat brain. Neurosci Lett 1996 Aug 2;213(2):103-6.
20. Pardridge WM, Oldendorf WH, Cancilla P, Frank HJ. Blood-brain barrier: interface between internal medicine and the brain. Ann Intern Med 1986 Jul;105(1):82-95.
21. Zhao WQ, Chen H, Quon MJ, Alkon DL. Insulin and the insulin receptor in experimental models of learning and memory. Eur J Pharmacol 2004 Apr 19;490(1-3):71-81.
22. Nitsch R, Hoyer S. Local action of the diabetogenic drug, streptozotocin, on glucose and energy metabolism in rat brain cortex. Neurosci Lett 1991 Jul 22;128(2):199-202.
23. Ding A, Nemeth G, Hoyer S. Age influences abnormalities in striatal dopamine metabolism during and after transient forebrain ischemia. J Neural Transm Park Dis Dement Sect 1992;4(3):213-25.
24. Lannert H, Hoyer S. Intracerebroventricular administration of streptozotocin causes long-term diminutions in learning and memory abilities and in cerebral energy metabolism in adult rats. Behav Neurosci 1998 Oct;112(5):1199-208.
25. Salkovic-Petrisic M, Hoyer S. Central insulin resistance as a trigger for sporadic Alzheimer-like pathology: an experimental approach. J Neural Transm Suppl 2007;72:217-33.
26. Machicao F, Muhlbacher C, Haring H. Inositol phospho-oligosaccharides from a dialysate (Actovegin) obtained from blood mimic the effect of lipogenesis glucose transport and lipolysis in rat adipocytes. Akt Endokr Stoffw 1989;10:111.
27. Kellerer M, Machicao F, Berti L, Sixt B, Mushack J, Seffer E, et al. Inositol phosphooligosaccharides from rat fibroblasts and adipocytes stimulate 3-O-methylglucose transport. Biochem J 1993 Nov 1;295 (Pt 3):699-704.
28. Reichel H, Weiss C, Leichtweiss HP. The effects of a blood extract on the oxygen uptake of isolated artificially perfused kidneys and skeletal muscles in rats. Arzneim-Forsch 1965;15(756):757.
29. de Groot H, Brecht M, Machicao F. Evidence for a factor protective against hypoxic liver parenchymal cell injury in a protein-free blood extract. Res Commun Chem Pathol Pharmacol Apr. 1990;68(1):125-8.
30. Hoyer S, Betz K. Elimination of the delayed postischemic energy deficit in cerebral cortex and hippocampus of aged rats with a dried, deproteinized blood extract (Actovegin). Arch Gerontol Geriatr 1989 Sep;9(2):181-92.
31. Murakami K, Shimizu T, Irie K. Formation of the 42-mer amyloid beta radical and the therapeutic role of superoxide dismutase in Alzheimer's disease. J Amino Acids 2011;2011:654207.
32. Zhang XH, Yu HL, Xiao R, Xiang L, Li L, Ma WW, et al. Neurotoxicity of betaamyloid peptide 31-35 and 25-35 to cultured rat cortical neurons. Zhonghua Yu Fang Yi Xue Za Zhi 2009 Dec;43(12): 1081-5.
33. Pike CJ, Walencewicz-Wasserman AJ, Kosmoski J, Cribbs DH, Glabe CG, Cotman CW Structure-activity analyses of beta-amyloid peptides: contributions of the beta 25-35 region to aggregation and neurotoxicity. J Neurochem 1995 Jan;64(1): 253-65.
34. Millucci L, Raggiaschi R, Franceschini D, Terstappen G, Santucci A. Rapid aggregation and assembly in aqueous solution of A beta (25-35) peptide. J Biosci 2009 Jun;34(2):293-303.
35. Trubetskaya VV, Stepanichev MY, Onufriev MV, Lazareva NA, Markevich VA, Gulyaeva NV. Administration of aggregated beta-amyloid peptide (25-35) induces changes in long-term potentiation in the hippocampus in vivo. Neurosci Behav Physiol 2003 Feb;33(2):95-8.
36. Klementiev B, Novikova T, Novitskaya V, Walmod PS, Dmytriyeva O, Pakkenberg B, et al. A neural cell adhesion molecule-derived peptide reduces neuropathological signs and cognitive impairment induced by Abeta25-35. Neuroscience 2007 Mar 2; 145(1):209-24.
37. Kubo T, Nishimura S, Kumagae Y, Kaneko I. In vivo conversion of racemized beta-amyloid ([D-Ser 26]A beta 1-40) to truncated and toxic fragments ([D-Ser 26] A beta 25-35/40) and fragment presence in the brains of Alzheimer's patients. J Neurosci Res 2002 Nov 1;70(3):474-83.
38. Meffert MK, Chang JM, Wiltgen BJ, Fanselow MS, Baltimore D. NF-kappa B functions in synaptic signaling and behavior. Nat Neurosci 2003 Oct;6(10):1072-8.
39. Kaltschmidt B, Kaltschmidt C. NF-kappa B in the nervous system. Cold Spring Harb Perspect Biol 2009 Sep;1(3):a001271.
40. Kaltschmidt B, Uherek M, Wellmann H, Volk B, Kaltschmidt C. Inhibition of NF-kappa B potentiates amyloid beta-mediated neuronal apoptosis. Proc Natl Acad Sci USA 1999 Aug 3;96(16):9409-14.
41. Meunier A, Latremoliere A, Dominguez E, Mauborgne A, Philippe S, Hamon M, et al. Lentiviral-mediated targeted NF-kappa B blockade in dorsal spinal cord glia attenuates sciatic nerve injury-induced neuropathic pain in the rat. Mol Ther 2007 Apr;15(4):687-97.
42. Mattson MP, Meffert MK. Roles for NF-kappa B in nerve cell survival, plasticity, and disease. Cell Death Differ 2006 May;13(5):852-60.
43. Tamatani M, Che YH, Matsuzaki H, Ogawa S, Okado H, Miyake S, et al. Tumor necrosis factor induces Bcl-2 and Bcl-x expression through NFkappa B activation in primary hippocampal neurons. J Biol Chem 1999 Mar 26;274(13):8531-8.
44. Kenchappa P, Yadav A, Singh G, Nandana S, Banerjee K. Rescue of TNFalpha-inhibited neuronal cells by IGF-1 involves Akt and c-Jun N-terminal kinases. J Neurosci Res 2004 May 15;76(4):466-74.
45. Waetzig GH, Rosenstiel P, Arlt A, Till A, Brautigam K, Schafer H, et al. Soluble tumor necrosis factor (TNF) receptor-1 induces apoptosis via reverse TNF signaling and autocrine transforming growth factor-beta1. FASEB J 2005 Jan;19(1):91-3.
46. Nawashiro H, Tasaki K, Ruetzler CA, Hallenbeck JM. TNF-alpha pretreatment induces protective effects against focal cerebral ischemia in mice. J Cereb Blood Flow Metab 1997 May;17(5):483-90.
47. Bruce AJ, Boling W, Kindy MS, Peschon J, Kraemer PJ, Carpenter MK, et al. Altered neuronal and microglial responses to excitotoxic and ischemic brain injury in mice lacking TNF receptors. Nat Med 1996 Jul;2(7):788-94.
48. Arnett HA, Mason J, Marino M, Suzuki K, Matsushima GK, Ting JP. TNF alpha promotes proliferation of oligodendrocyte progenitors and remyelination. Nat Neurosci 2001 Nov;4(11):1116-22.
49. Barger SW, Horster D, Furukawa K, Goodman Y, Krieglstein J, Mattson MP. Tumor necrosis factors alpha and beta protect neurons against amyloid betapeptide toxicity: evidence for involvement of a kappa B-binding factor and attenuation of peroxide and Ca2+ accumulation. Proc Natl Acad Sci USA 1995 Sep 26;92(20):9328-32.
50. Kaltschmidt C, Kaltschmidt B, Neumann H, Wekerle H, Baeuerle PA. Constitutive NF-kappa B activity in neurons. Mol Cell Biol 1994 Jun;14(6):3981-92.
51. Pieper AA, Verma A, Zhang J, Snyder SH. Poly (ADP-ribose) polymerase, nitric oxide and cell death. Trends Pharmacol Sci 1999 Apr;20(4):171-81.
52. Eliasson MJ, Sampei K, Mandir AS, Hurn PD, Traystman RJ, Bao J, et al. Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat Med 1997 Oct;3(10):1089-95.
53. Burkart V, Wang ZQ, Radons J, Heller B, Herceg Z, Stingl L, et al. Mice lacking the poly(ADP-ribose) polymerase gene are resistant to pancreatic beta-cell destruction and diabetes development induced by streptozocin. Nat Med 1999 Mar;5(3):314-9.
54. Pieper AA, Brat DJ, Krug DK, Watkins CC, Gupta A, Blackshaw S, et al. Poly(ADP-ribose) polymerase-deficient mice are protected from streptozotocin-induced diabetes. Proc Natl Acad Sci USA 1999 Mar 16;96(6):3059-64.
55. Garcia SF, Virag L, Jagtap P, Szabo E, Mabley JG, Liaudet L, et al. Diabetic endothelial dysfunction: the role of poly(ADP-ribose) polymerase activation. Nat Med 2001 Jan;7(1):108-13.
56. Ilnytska O, Lyzogubov VV, Stevens MJ, Drel VR, Mashtalir N, Pacher P, et al. Poly(ADP-ribose) polymerase inhibition alleviates experimental diabetic sensory neuropathy. Diabetes 2006 Jun;55(6): 1686-94.
Комментарии
ПРАКТИКА ПЕДИАТРА



