Конференция «Эпилептические энцефалопатии.
Когнитивные дезинтеграции» Объединения врачей эпилептологов и пациентов.
В 2011 году по инициативе Международного Бюро по эпилепсии (International Bureau for Epilepsy – IBE) и при поддержке Международной Лиги по Борьбе с Эпилепсией (ILAE) был утвержден Европейский День Эпилепсии, который отмечается 14 февраля – в день святого Валентина – покровителя больных эпилепсией в европейских странах. В России покровитель больных эпилепсией – святой Трифон, чей праздник так же отмечается 14 февраля.
В 2012 году этот праздник отмечался уже во второй раз. Основная тема праздника – «Бремя эпилепсии», идея которой заключена в необходимости освобождения больных эпилепсией от бремени социальной стигматизации и дискриминации, связанных с их заболеванием. Празднование в России проходило в два этапа.
В первый день празднования 17 февраля 2012 года – в концертном зале Московского Дома композиторов проводился благотворительный праздничный концерт для врачей, пациентов и их близких. Он был организован Фондом помощи больным эпилепсией «Содружество» при участии Объединения врачей-эпилептологов и пациентов. В концерте перед собравшимися слушателями (около 400 человек) выступили молодые талантливые музыканты – лауреаты международных и всероссийских конкурсов, обладатели престижных премий и наград в области музыкального искусства – юношеского симфонического оркестра России им. Л.В. Николаева (художественный руководитель и главный дирижер Василий Валитов).
Во второй день празднования – 27 февраля 2012 года – Объединением врачей-эпилептологов и пациентов для врачей была организована Конференция «Эпилептические энцефалопатии. Когнитивные дезинтеграции». Конференция традиционно проводилась в гостинице при Даниловском монастыре, где создается особая атмосфера при обсуждении аспектов «священной болезни».
В целом в работе Конференции приняли участие 140 делегатов.
Конференция проводилась при спонсорской поддержке компаний: генерального спонсора – Санофиавентис груп, главных спонсоров – ЮСБ фарма и Эйсай, спонсора – Валента Фарм, а так же при участии Региональной Общественной Организации «Общество специалистов по нервно-мышечным болезням», Фонда «Содружество».

Леонид Ростиславович Зенков на Конференции «Инновации в эпилептологии» 21 октября 2011 года представлял доклад «Необычные формы симптоматических фокальных эпилепсий».
Собравшиеся на Конференции почтили минутой молчания память скоропостижно скончавшегося 26 февраля 2012 года великого русского ученого Леонида Ростиславовича Зенкова, доктора медицинских наук, профессора, главного научного сотрудника Лаборатории клинико-электрофизиологических исследований при кафедре нервных болезней лечебного факультета Первого Московского Государственного медицинского университета им. И.М. Сеченова.

Андрей Сергеевич Петрухин – главный детский невролог России, президент Объединения врачей-эпилептологов и пациентов выступает с приветственным словом перед собравшимися на концерт.
Конференцию по традиции открыл член-корреспондент РАМН, Заслуженны деятель науки РФ, почетный президент Объединения врачей-эпилептологов и пациентов, д.м.н., профессор Московского государственного медикостоматологического университета В.А. Карлов с фундаментальным докладом «Попытка классификации парциальных эпилептических энцефалопатий». Известно, что эпилептический процесс при хронической текущей эпилепсии имеет проградиентную морфологическую основу, что в значительной мере связано с повторными эпилептическими припадками и сопровождающими их аноксиишемическими изменениями в очаге и в наиболее уязвимых структурах мозга.На этом основании было сформулировано положение об эпилептической энцефалопатии как клинико-морфологическом понятии. Владимир Алексеевич предложил выделять с практических позиций два типа эпилептической энцефалопатий: с клиническим проявлением эпилептических приступов, речевыми, когнитивными и поведенческими расстройствами; без эпилептических припадков, с наличием только речевых, когнитивных и поведенческих расстройств. Если при эпилептической энцефалопатии первого типа диагноз очевиден по клиническим проявлениям заболевания, то при энцефалопатии второго типа он может быть только заподозрен и подтвержден ЭЭГ, в ряде случаев только с применением полиграфии ночного сна. Далее в своем докладе В.А. Карлов представил Классификацию эпилептических энцефалопатий, иллюстрированную значительным количеством электроэнцефалограмм пациентов. В.А. Карлов отметил проблемы, связанные с терапевтическими подходами. В лечении эпилептических энцефалопатий сочетаются два взаимоисключающих принципа: немедленное начало и наоборот, не торопиться с началом. Основное значение имеют, при наличии соответствующей клиники, данные ЭЭГ: регистрация типичной эпилептической, а тем более гигантскоамплитудной остромедленноволновой активности, как правило, требует немедленного старта терапии. Средством выбора являются АЭП, обладающие широтой действия: вальпроаты, ламотриджин и леветирацетам, при электрическом статусе сна – этосуксимид или их комбинации. В целом, при регистрации доброкачественных разрядов детского возраста, как правило, решение принимается после дополнительных обследований, прежде всего психологического тестирования, либо оценки последующей динамики заболевания.

Андрей Сергеевич Петрухин, Владимир Алексеевич Карлов и Константин Юрьевич Мухин – председатели симпозиума.
Главный детский невролог РФ, президент «Объединения врачейэпилептологов и пациентов», д.м.н., профессор А.С. Петрухин сделал общий доклад «Нейропсихиатрические аспекты эпилепсии у детей». Эпилепсия представляет собой заболевание, стоящее на стыке неврологии и психиатрии. Это связано не только с тем, что иктальные состояния могут проявляться психическими нарушениями, но так же и с фактом, что в перииктальных периодах, а так же в межприступном периоде пациента могут беспокоить изменения когнитивных функций, а так же аффективно-личностные нарушения, что объективно нашло свое отражение в современном определении эпилепсии. В целом, у 30-60% больных с эпилепсией наблюдаются нейропсихиатрические проблемы. Психосоциальные проблемы для пациентов часто выходят на первый план, включая случаи, когда контроль над приступами еще не достигнут, а так же и по достижению контроля над ними. Существуют определенные различия между нарушениями когнитивных и поведенческих функций в детском и во взрослом возрастах. Особенностью эпилепсии у детей является влияние эпилептических приступов и антиэпиептических препаратов (АЭП) на развитие структур центральной нервной системы и на формирование высших психических функций (ВПФ), обеспечивающих адаптацию организма ребёнка к условиям внешней среды. Это отражается в более выраженных изменениях личностной сферы и функций, составляющих основу когнитивной деятельности (внимание, гнозис, память, мышление). В свою очередь, пластичность психических процессов у детей обусловливает возможность компенсации нарушений при направленной коррекции. Кроме того, у детей встречаются особые состояния – ранние злокачественные эпилептические энцефалопатии и когнитивные дезинтеграции. В целом, развитие описываемых нарушений – полиэтиологично. Имеет значение так же и назначение АЭП в моно- или политерапии (ятрогения), а так же – несвоевременное назначение АЭП. На фоне проводимой антиэпилептической терапии когнитивные расстройства могут иметь чаще всего дозозависимый транзиторный или более продолжительный характер, однако могут развиваться и хронические побочные эффекты АЭП с перманентным, в некоторых случаях – прогрессирующим, нарушением когнитивных функций. В большинстве случаев транзиторные когнитивные нарушения трансформируются в перманентные и даже прогрессирующие при длительном течении эпилепсии, резистентной к антиэпилептической терапии. Описываемые расстройства могут приводить к социальной дезадаптации и стигматизации, которые, в свою очередь, оказывают негативное влияние на нарушения в высшей психической сфере. Может создаваться так называемый «порочный круг». В настоящее время известно, что не у всех больных с эпилепсией развиваются когнитивные и аффективно-личностные нарушения, очевидно, благодаря своевременной и точной диагностике и современной антиэпилептическиой терапии, а также – возможностям коррекции нарушений.
Психические расстройства при эпилепсии у детей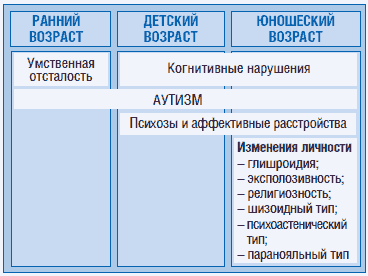
Этиология и характер психических расстройств при эпилепсии в младенческом и раннем детском возрасте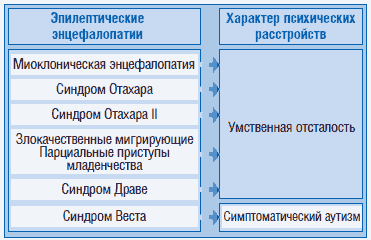
Нейропсихиатрические проблемы и психические нарушения в детском возрасте (дошкольном и младшем школьном)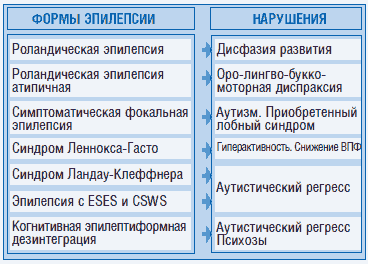
Нейропсихиатрические проблемы в юношеском возрасте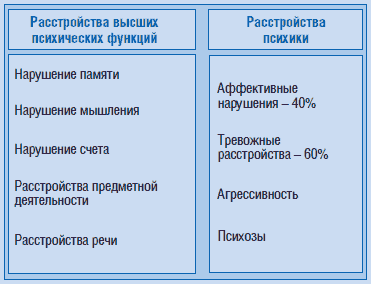
Одной из наиболее часто встречающихся когнитивных проблем у больных с эпилепсией является нарушение памяти. Пациентов с эпилепсией могут так же беспокоить проблемы, связанные с нарушением внимания. В частности – в рамках синдрома СДВГ (синдрома гиперактивности с дефицитом внимания). Нарушение внимания может беспокоить больных с абсансными формами эпилепсии. Речевые проблемы (чаще в чтении и письме) могут возникать у больных с эпилепсией без нарушений интеллекта. Описаны эпилептические синдромы со специфическими речевыми нарушениями. Например, синдром или афазия Ландау-Клеффнера. Диагностика данного синдрома бывает затруднена в связи с тем, что у отдельных пациентов эпилептические приступы не развиваются. У некоторых больных припадки, наоборот, могут предшествовать афатическим нарушениям.
Группа эпилептических синдромов, для которых патогномонично снижение интеллекта, – ранние злокачественные энцефалопатии у детей, у которых приступы манифестируют в большинстве случаев на первом году жизни. У большинства выживших пациентов интеллектуальные нарушения либо манифестирующие практически одновременно с дебютом приступов или ассоциирующиеся с задержкой психического развития, являются одним из основных симптомов болезни, в дальнейшем интеллектуальные нарушения могут приобретать характер плато. Нарушения интеллекта отмечаются у больных с такими заболеваниями, как синдром Кожевникова-Расмуссена, прогрессирующие формы эпилепсии с миоклонусом. У детей с электрическом статусом медленно-волнового сна интеллектуальный дефицит, который отмечается даже в отсутствие приступов, может регрессировать по мере редукции эпилептиформной активности на ЭЭГ. В случае если терапия начинается несвоевременно, интеллектуальные нарушения могут приобретать стойкий характер. Однако в настоящее время убедительно показано, что и в случаях, когда терапия назначалась вовремя, в дальнейшем все равно может сохраняться интеллектуальный дефицит.
Многие исследователи рекомендуют для улучшения когнитивных функций как можно раньше обеспечить надежный контроль над приступами, избегать назначения препаратов «старых групп» и назначать с осторожностью топирамат. В отдельных случаях рекомендован пересмотр антиэпилептической терапии: уменьшение числа препаратов в комбинации, замена препаратов, применение препаратов с отсутствием негативного влияния на когнитивные функции действием –вальпроата, окскарбазепина, ламотриджина, габапентина, леветирацетама, лакосамида. Значительную роль в коррекции дефицита высшей психической сферы играет направленная психологическая коррекция. Необходимо учитывать возможности назначения симптоматической терапии, в частности препарата ноотропного ряда – пантокальцина.
Второй доклад профессора А.С. Петрухина был посвящен «Инновациям в лечении эпилепсии в России». Поиск возможностей для увеличения эффективности АЭП включает: синтез новых лекарственных формул или модификацию уже синтезированных молекул, определение новых показаний к назначению, поиск новых лекарственных форм, новых рациональных комбинаций препаратов и другие. Современные методы решения проблемы побочных эффектов АЭП включают: профилактику (назначение АЭП с учетом общего соматического и психического здоровья пациента и его ближайших родственников), коррекцию побочных эффектов, мониторинг состояния: своевременные обследования, консультации специалистов.
На российском фармацевтическом рынке широко представлены новые формулы АЭП с новыми механизмами действия. В России в 2010 был зарегистрирован лакосамид (вимпат) для лечения фокальных эпилепсий у пациентов старше 16 лет в комбинированной терапии. У лакосамида открыт новый противоэпилептический механизм. Препарат избирательно усиливает медленную инактивацию натриевых каналов без влияния на быструю инактивацию, в результате чего происходит: стабилизация гипервозбудимых нейрональных мембран, нормализация порогов активации и ингибирование повторяющихся нейрональных разрядов. В настоящее время представлена как таблетированная форма лакосамида, так и внутривенный раствор, который является альтернативой для пациентов, временно неспособных принимать препарат per os. В настоящее время на территории РФ на стадии регистрации находятся и другие новейшие противоэпилептические препараты: зонисамид (зонегран) в капсулах в качестве дополнительного лекарственного препарата у взрослых при лечении парциальных эпилептических припадков с и без вторичной генерализации и руфинамид (иновелон) в таблетках в качестве дополнительного лекарственного препарата при лечении эпилептических припадков, ассоциированных с синдромом Леннокса – Гасто у пациентов старше 4 лет. У зонисамида механизм действия – комплексный, однако до конца не раскрыт. Предполагается, что он воздействует на потенциалзависимые натриевые и кальциевые каналы, а так же снижает активность нейронов посредством гамма-аминомасляной кислоты. Руфинамид является модулятором натиевых каналов. В США был зарегистрирован в 2004г., в Европе – 2007г. В клинических исследованиях убедительно доказана его эффективность в терапии дропатак (уменьшает их на 42,5%, а эффект становится заметным уже в первые 2 недели). Недавно было показано, что руфинамид может быть эффективным в комплексной терапии эпилептических энцефалопатий и эпилептических синдромов, помимо синдрома Леннокса-Гасто, – синдрома Драве, злокачественных мигрирующих парциальных приступах младенчества и других (G.Coppola, 2010).
Одним из методов поиска новых лекарств является – модификация уже синтезированных молекул.
В России в настоящее время проходит государственная регистрация новейшего препарата перампанел (файкомпа) в таблетках, который является селективным антагонистом AMPA-рецептора глутамата для лечения фокальных эпилепсий, резистентных к предыдущим протовоэпилептическим препаратам. Регистрируется так же препарат эсликарбазепин (эксалиеф) в таблетках для лечения фокальных приступов с и без вторичной генерализации. Препарат уже зарегистрирован во многих европейских странах и успешно применяется в соответствии с показаниями и в особенности при проблемах с переносимостью карбамезепина и окскарбазепина.
Утверждены новые показания к назначению отдельных АЭП. Например для леветирацетама (кеппры) раствора для приема внутрь (1 мл содержит 100 мг леветирацетама). Препарат показан в качестве монотерапии (препарат первого выбора) при лечении парциальных припадков с вторичной генерализацией или без таковой у взрослых и подростков старше 16 лет с вновь диагностированной эпилепсией и в составе комплексной терапии: при лечении парциальных припадков с вторичной генерализацией или без таковой у взрослых и подростков старше 1 месяца, страдающих эпилепсией; миоклонических судорог у взрослых и подростков старше 12 лет, страдающих ювенильной миоклонической эпилепсией; первичногенерализованных судорожных (тонико-клонических) припадков у взрослых и подростков старше 12 лет, страдающих идиопатической генерализованной эпилепсией.
Как уже сообщалось, еще одна возможность для улучшения показателей эффективности и переносимости антиэпилептической терапии – создание новых лекарственных форм препаратов, уже существующих в определенных лекарственных формах, чаще всего – в таблетках. Например, в России была зарегистрирована новая форма вальпроата с пролонгированным действием – Депакин хроносфера. Ее разработка была продиктована одной из основных проблем в лечении эпилепсии – недостаточным соблюдением режима приема препарата, особенно – при 3-х кратном приеме, что является основной причиной срыва ремиссии и неэффективного контроля припадков. У большого числа пациентов (преимущественно детского возраста и лиц с нарушением самоконтроля) имеются трудности проглатывания таблеток или капсул с активным веществом. Лекарственная форма микрогранулы (Депакин хроносфера) представляет собой безвкусные сферические гранулы диаметром ≈400 микрон, расфасованные в пакетики по 100, 250, 500, 750 и 1000 мг, что обеспечивает достаточно широкий диапазон дозировок с возможностью их еще более тщательного подбора. Препарат насыпается на поверхность мягкой пищи или напитка комнатной температуры и потребляется вместе с ними. Можно также просто высыпать порошок в рот и запить его водой. Причем, показано отсутствие взаимодействия пищи с Депакином хроносфера. Препарат полностью биоэквивалентен другим пролонгированным формам Депакина. Дополнительными преимуществами вальпроевой кислоты являются: прогнозируемый профиль безопасности, предсказуемость появления тех или иных нежелательных явлений, хорошая корреляция клинических и нейрофизиологических эффектов, отсутствие парадоксального утяжеления припадков, что было показано в многочисленных зарубежных и российских исследованиях. Одно из последних российских исследований, посвященное аггравации эпилепсии, – практически эпидемиологическое по количеству вошедших в него пациентов – Проваторовой М.А., 2011, так же корреспондировало со всем вышесказанным. Таким образом, Депакин хроносфера представляет собой уникальную пролонгированну лекарственную форму вальпроата для взрослых и детей удобен в применении (упаковка в форме пакетиков), не имеет вкуса и запаха, позволяет упростить режим приема и может эффективно и безопасно использоваться в режиме приема 1 раз в сутки, что значительно улучшает комплаенс, и, как следствие, – результаты проводимой терапии.
В завершении А.С. Петрухин еще раз подчеркнул, что поиск разумного баланса между эффективностью проводимой антиэпилептической терапии и ее безопасностью – залог высокого комплаенса и сохранения качества жизни пациента с эпилепсией.
Руководитель Института детской неврологии и эпилепсии имени Святителя Луки, д.м.н., профессор К.Ю. Мухин сделал сообщение «Когнитивная эпилептиформная дезинтеграция. Дефиниция, диагностика, терапевтическая тактика».
Когнитивная эпилептиформная дезинтеграция (КЭД) – симптомокомплекс приобретенных нарушений высших психических функций у детей, ассоциированный с выраженной эпилептиформной активностью на ЭЭГ, при отсутствии эпилептических приступов. При этом допускается возможность единичных эпилептических приступов в анамнезе (К.Ю. Мухин, 2009; Gobbi, 2002).
R.Tuchman & I.Rapin в 1997 году представили спектр различных вариантов «когнитивной дезинтеграции» в педиатрической практике и обратили внимание на частое сочетание дезинтегративных расстройств с эпилептиформной активностью на ЭЭГ. Позднее G. Gobbi и соавт. (2002) на международном конгрессе в Лиссабоне ввели термин «когнитивная эпилептиформная дезинтеграция» – приобретенное нарушение когнитивных функций в результате постоянной продолженной эпилептиформной активности на ЭЭГ. При этом подразумевается отсутствие у пациентов эпилептических приступов или наличие единичных приступов в анамнезе. В русскоязычной литературе этот термин был впервые предложен нами (К.Ю. Мухин, 2004).
Термин КЭД не идентичен понятию «эпилептическая энцефалопатия», так как в последнем случае когнитивные нарушения могут быть обусловлены частыми эпилептическими приступами, как например, при синдромах Веста, Драве, Леннокса-Гасто и многих других. При КЭД глобальные приобретенные нарушения когнитивных, речевых функций и поведения у детей обусловлены разрывом нейрональных связей развивающегося мозга в результате постоянной продолженной эпилептиформной активности в условиях «врожденного нарушения процессов созревания мозга», которое, по-видимому, генетически детерминировано и нередко сочетается с внутриутробной гипоксией. КЭД объединяет группу различных синдромов, связанных общим патогенезом.
Варианты когнитивной эпилептиформной дезинтеграции и схожих синдромов
|
Целью терапии антиэпилептическими препаратами (АЭП) (даже при отсутствии эпилептических приступов) или стероидными гормонами при КЭД является блокирование постоянной продолженной эпилептиформной активности на ЭЭГ, что позволяет сократить период существования этой активности и тем самым уменьшить выраженность когнитивного дефекта в исходе заболевания (Л.Р. Зенков, 2004; К.Ю. Мухин и соавт., 2005; Duarte и соавт., 2007). Показаниями к назначению лечения являются:
- Наличие эпилептических приступов.
- Выраженные нарушения когнитивных функций, речи или поведения в сочетании с постоянной продолженной эпилептиформной активностью на ЭЭГ.
- Нарастание индекса эпилептиформной активности в сочетании с углублением когнитивных нарушений в динамике.
При наличии выраженных расстройств высших психических функций с единичными эпилептиформными паттернами, а также у больных с минимальными нарушениями когнитивных функций и высоким индексом эпилептиформной активности, как правило, рекомендуется воздержаться от назначения терапии АЭП.
Наиболее эффективными АЭП в лечении КЭД признаны: вальпроаты, сукцинимиды, сультиам, топирамат, леветирацетам и бензодиазепины. Препараты группы карбамазепина, окскарбазепин, прегабалин, барбитураты, фенитоин – противопоказаны ввиду высокого риска аггравации приступов и диффузных эпилептиформных паттернов на ЭЭГ (В.А. Карлов, 2006; Doose & Baier, 1989). Рекомендуется воздержаться от назначения седативных препаратов, которые, снижая уровень бодрствования и повышая сонливость, также могут способствовать аггравации эпилептиформных нарушений.
При отсутствии эпилептических приступов у больных КЭД, лечение может начинаться с вальпроатов (депакин, конвулекс), сукцинимидов или сультиама. При наличии эпилептических приступов стартовая терапия осуществляется только с вальпроатов. Резервными препаратами в монои политерапии являются топирамат (топамакс) и леветирацетам (кеппра). Бензодиазепины (клобазам) – резервные препараты, преимущественно, в комбинированной терапии (Fejerman & Caraballo, 2007). Принципиально важно, что у всех больных КЭД, в конечном итоге, достигается клиническая ремиссия – полное купирование приступов. Однако с исчезновением приступов нейропсихологический дефицит уменьшается, но не исчезает. Необходимо добиться блокирования продолженной эпилептиформной активности на ЭЭГ, что у многих пациентов коррелирует с улучшением когнитивных функций.
По мнению большинства исследователей, наибольшей эффективностью в отношении улучшения высших психических функций у больных КЭД обладают кортикостероидные гормоны (Beaumanoir и соавт., 1995; Fejerman & Caraballo, 2007). Предложены различные схемы применения кортикостероидов: дексаметазона, гидрокортизона, преднизолона. Мы рекомендуем применение гидрокортизона (Sanofi-аventis) перорально. Назначение кортикостероидов улучшает когнитивные функции не только за счет уменьшения выраженности эпилептиформной активности, но и в результате «ускорения процессов созревания мозга» (Doose и соавт., 2000; Sutula, 2004). Предпочтение отдается именно кортикостероидам, а не синтетическим аналогам АКТГ (нативный АКТГ в настоящее время практически нигде не применяется). Все авторы рекомендуют длительные курсы кортикостероидов. Ограничивает назначение данных препаратов высокая частота побочных эффектов, в том числе, серьезных. Кортикостероиды назначаются вместе с препаратами калия. Начинать лечение необходимо в условиях стационара.
Профессор Е.Д. Белоусова (Московский НИИ педиатрии и детской хирургии Минздравсоцразвития РФ») с соавтором доклада – к.м.н. А.Ю. Ермаковым сделала доклад «Эпилептическая энцефалопатия с продолженной сайк-волновой активностью во сне». Эпилепсия с электрическим эпилептическим статусом в фазу медленного сна или энцефалопатия с электрическим эпилептическим статусом в фазу медленного сна является частично обратимой возрастзависимой эпилептической энцефалопатией, для которой характерна триада симптомов: продолженная спайк-волновая активность во сне (синоним – электрический эпилептический статус в фазу медленного сна), cудороги, нейропсихологические нарушения. Некоторые авторы считают, что понятия «электрический эпилептический статус в фазу медленного сна» (ESES – cокр. от англ. electrical status epilepticus in sleep) и «продолженная спайк-волновая активность во сне» (CSWS – сокр. от англ. continuous spikes and waves during sleep) являются полными синонимами. Некоторые авторы считают, что «электрический эпилептический статус в фазу медленного сна» описывает только изменения на ЭЭГ, а «продолженная спайкволновая активность во сне» представляет собой комбинацию ЭЭГ аномалий и нейропсихологических нарушений. Считается, что частота эпилептической энцефалопатии с CSWS составляет 0,5% всех эпилепсий у детей. Этиология до сих пор считается недостаточно изученной. Начало эпилептических приступов приходится на возраст от 2-х месяцев до 12 лет (с пиком дебюта эпилепсии от 4 до 5 лет). CSWS начинается через 1–2 года после начала приступов (от 3 до 14 лет, пик в 8 лет). Определяется стадийность течения заболевания, но иногда эволюция по стадиям не определяется. Характер нейропсихиатрических нарушений связан с преимущественной локализацией эпилептиформной активности. Активность преимущественно лобной локализации вызывает когнитивные нарушения и нарушения конструктивной деятельности до развития речевых нарушений. Формируется так называемый «лобный» тип психики – расторможенность, гиперактивность, агрессивность, раздражительность, черты аутизма, ажитация в сочетании с нарушениями памяти и внимания. Эпилептиформная активность преимущественно височной локализации вызывает, прежде всего, речевые нарушения, преимущественно моторную афазию (в отличии от вербальной агнозии при синдроме ЛандауКлеффнера). Первоначально ESES описывался как диффузный паттерн. Но в последние годы все чаще стали писать о том, что разряды во сне могут быть асимметричными, унилатеральными или преимущественно фокальными и это не служит критерием исключения СSWS. По данным разных авторов спайк-волновый индекс при этом синдроме колеблется от 25 до 90%, а некоторые авторы и вовсе ограничиваются фактом «значительной активации разрядов во сне».
Приступы могут оказаться как чувствительными, так и не чувствительными к лечению. Комбинация вальпроата и этосуксимида считается оптимальной. Авторы доклада особо отметили, что предпочтительным является применение вальпроатов в виде пролонгированной формы – Депакин хроносфера, дающей дополнительные свойства эффективности и безопасности у детей вследствие того, что: концентрация препарата в плазме стабильно поддерживается на верхней границе терапевтического диапазона при однократном или двукратном приеме; пища не влияет на биодоступность вальпроевой кислоты в виде микрогранул; не возникает трудностей при проглатывании; отмечается удобство в применении – возможен однократный прием. Карбамазепин может аггравировать энцефалографический статус. Можно попробовать применение леветирацетама, топирамата и ламотриджина. Об эффективности применения леветирацетама сообщают многие зарубежные и отечественные авторы. Atkins M. и соавт., 2011, сообщили о своих наблюдениях 20 пациентов, которым назначался леветирацетам 45–50 мг на кг веса как дополнительный препарат. Оценка эффективности производилась через 18 месяцев применения. Положительные изменения на ЭЭГ были выявлены у 11 пациентов. Кроме того, имеются данные о том, что электрический статус «чувствителен» к бензодиазепинам (клобазам), сультиаму (осполот) и АКТГ или преднизолону.
В течение времени происходит уменьшение активности – разряды становятся короче и реже, более фрагментированными. Становятся более заметными нормальные паттерны сна. Могут длительно сохраняться фокальные комплексы острая-медленная волна. Нормализация ЭЭГ развивается через годы – в среднем около 11 лет, иногда после 15 лет. Приступы прекращаются в возрасте от 10 до 15 лет. Степень выраженности нейропсихиатрических нарушений зависит от возраста развития CSWS и продолжительности ее существования. Большинство пациентов не возвращаются к нормальному уровню внимания и речевых функций. Лишь у четверти пациентов отмечается приемлемый уровень интеллекта, внимания и речи (нормальный интеллект и статус до начала эпилепсии и короткая продолжительность CSWS).
Н.А. Ермоленко (д.м.н., Воронежская государственная медицинская академия им. Н.Н. Бурденко, вице-президент НП «Объединение врачей-эпилептологов и пациентов) сделала доклад «Когнитивные нарушения при роландической эпилепсии».
Роландическая эпилепсия (РЭ) является одной из наиболее частых форм эпилепсии детского возраста, которая составляет по данным C.P. Panayiotopoulos около 15% среди детей с эпилепсией в возрасте до 15–16 лет, с исключением неонатального периода. В 1989 году был выдвинут один из критериев РЭ – отсутствие интеллектуального или нейропсихологического дефицита. Обнаружение того факта, что дети с РЭ часто имеют когнитивный дефицит и проблемы поведения, растянулось на 30–40 лет, вопреки очевидным изменениям в когнитивной сфере у пациентов, упомянутым при первых описаниях синдрома. Только в 2000 году T.W. Deonna с соавторами опубликовали результаты длительного проспективного исследования, в котором была определена «приобретенная пролонгированная обратимая дисфункция» связанная с эпилептиформной активностью на ЭЭГ детей с РЭ. В настоящее время доказано, что помимо эпилептических припадков РЭ сопровождается спектром нейропсихологических расстройств, из которых наиболее яркими являются нарушения речи и языка как следствие глоссо-, оро-, фаринголарингеальной диспраксии, дизартрии, дезартикуляции, речевой диспраксии, аграмматизма, сенсорной и моторной афазии, алексии, аграфии, возникая в одних случаях до появления припадков, в других – совпадая с их возникновением и в третьих – появляясь в отдаленном катамнезе, когда диагноз эпилепсии снят из-за прекращения припадков, что позволяет рассматривать РЭ как минимальное проявление эпилептической энцефалопатии.
Нами проведено проспективное исследование в течение 5 лет пациентов с РЭ (n=44) в возрасте от 5 до 11 лет (средний возраст 7,3±1,6), целью которого было определение особенностей нейропсихологического дефицита и установление связи с длительностью и локализацией региональных эпилептиформных изменений, определение наиболее эффективных схем антиэпилептической терапии (АЭТ). Все дети, находившиеся под наблюдением по достижении школьного возраста, обучались в общеобразовательной школе. Однако при нейропсихологическом тестировании на момент обращения до назначения АЭТ (средний возраст 7,3±1,6) когнитивные функции были сохранными лишь у 11% (n=5) пациентов. У большинства детей выявлены различные нарушения в когнитивной сфере, главным образом связанные с слухоречевой памятью, вербальными функциями, оптико-моторной координацией и нарушением поведения, которые по достижении клиникоэлектроэнцефалографической ремиссии значительно уменьшались, но сохранялись на протяжении всего периода наблюдения за пациентами. Таким образом, последствиями фокальной эпилептиформной активности центро-темпоральных спайков на ЭЭГ при роландической эпилепсии являются временные когнитивные/поведенческие проблемы, следовательно, РЭ не являются «доброкачественными», как предполагалось ранее. В лечении этой категории пациентов необходимо добиваться не только клинической, но и электроэнцефалографической ремиссии, что требует длительного (не менее 36 месяцев) приема антиэпилептических препаратов (АЭП) не только в монотерапии, но и в комбинации двух или трех АЭП. Препаратами выбора являются вальпроаты в монотерапии (Депакин хроно, Депакин хроносфера) или в комбинации с этосуксимидом (суксилепом) и леветирацетамом (кеппрой).
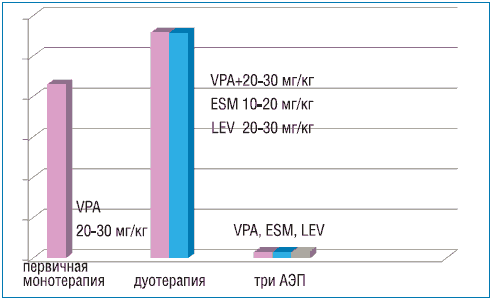
Варианты терапии пациентов с РЭ, достигших ремиссии (собственные данные Н.А. Ермоленко и соавт.)
Н.Ю. Перунова, д. м. н., председатель Совета НП «Эпилептологи Урала», консультант Областного детского центра эпилепсии и пароксизмальных состояний ОДКБ № 1 и Т.Р. Томенко, к.м.н., невролог-эпилептолог детской поликлиники Европейского медицинского центра УГМК (Екатеринбург) представили доклад «Клинико-энцефалографическая динамика эпилептических энцефалопатий с продолженной спайк-волновой активностью во сне».
Авторы привели данные публикаций: Smith M.C. и соавт., 2003; Rossi P.G. и соавт., 2004; Nickels K. и соавт., 2006; Tassinari C.A. и соавт., 2006; Van HirtumDas M. и соавт., 2006; Wang S. соавт., 2006; Praline J. и соавт., 2006; Scholtes F.B. и соавт., 2010; Garcнa-Peсas J.J., 2010; Liukkonen E. и соавт., 2010; Loddenkemper T. и соавт., 2011.
Детские эпилептические энцефалопатии с паттерном продолженной спайк-волновой активности во сне (CSWS) принято рассматривать как модель воздействия эпилептиформной активности на высшие психические функции. Такие энцефалопатии, как эпилептический электрический статус медленноволнового сна (ESES) и эпилептическая афазия Ландау-Клеффнера, считаются отдельными формами единого континуума. Выраженность резидуального дефицита, формирующегося в исходе этих заболеваний, пропорциональна продолжительности активного периода. Критической длительностью активного периода эпилептических энцефалопатий с CSWS (ЭЭ с CSWS) предложено считать срок от 2 до 3 лет, после чего полноценное восстановление интеллектуальных функций невозможно.
В качестве отрицательных факторов прогноза ЭЭ с CSWS выделяют ранний возраст дебюта (до 3 лет), большую продолжительность активной фазы течения заболевания (более 3 лет), низкий уровень и длительное время диагностики, отсутствие «ответа» на терапию. Прогноз нарушений речи при эпилептических энцефалопатиях с CSWS считается неблагоприятным при дебюте заболевания в возрасте до 3 лет, сохранении речевых нарушений более 1 года. В течении ЭЭ с CSWS предложено выделять активный период, характеризующийся персистированием паттерна CSWS, и резидуальный период. Проспективные исследования, содержащие информацию о динамике течения ЭЭ с CSWS, немногочисленны. Анализируются данные по группам из 11–8 пациентов (детей, подростков, молодых взрослых), длительность наблюдения от 5 до 9 лет, максимально –16 лет.
Основными синдромами ЭЭ с CSWS являются наличие эпилептических припадков, расстройства поведения, когнитивные нарушения, нарушения речи, изменения на ЭЭГ.
Эпилептические припадки возникают у 80% пациентов, страдающих ЭЭ с CSWS. Назначение антиконвульсантов оказывается эффективным в отношении контроля припадков в 60% случаев, однако воздействие на эпилептиформную активность является недостаточным. В отдаленном периоде эпилептические припадки могут сохраняться у 14% пациентов.
Расстройства поведения в острой фазе ЭЭ с CSWS наблюдаются у 90% детей, однако при проспективном наблюдении у 75% из них отмечается постепенное снижение выраженности поведенческих нарушений.
Когнитивные нарушения в активном периоде ЭЭ с CSWS встречаются у 90% пациентов. Хорошее восстановление когнитивных функций возможно только у 10%, обучение в массовой школе – у 12%. Выраженный интеллектуальный дефицит в отдаленном периоде сохраняется у 18% подростков и молодых взрослых.
Полный регресс нарушений речи в исходе ЭЭ с CSWS возможен в 17–18% случаев, частичное восстановление нарушений речи – в 37–40%. У 50–60% пациентов речевые нарушения сохраняются в течение всей жизни.
Продолженная эпилептиформная активность с индексом более 50% – облигатный симптом ЭЭ с CSWS. В активном периоде эпилептиформная активность оценивается как диффузная в 40%, случаев, как диффузная с региональными преобладанием – в 60%. Клинические проявления ЭЭ с CSWS и характеристики эпилептиформной активности не всегда совпадают. Персистирование изменений на ЭЭГ при синдроме Ландау-Клеффнера регистрируется до 5 лет, при ESES – до 9 лет.
Авторы представили собственные данные проспективного наблюдения группы пациентов, страдающих ЭЭ с CSWS. Критериями включения в исследования были: регистрация по данным ЭЭГ-видеомониторинга сна паттерна CSWS, отсутствие структурных изменений мозга по данным МРТ; продолжительность наблюдения не менее 3 лет; проспективное клиническое наблюдение, повторное проведение ЭЭГвидеомониторинга.
В исследование вошли 15 пациентов (4 девочки и 11 мальчиков) не более чем через 6–7 месяцев после дебюта заболевания. Возраст пациентов на момент последнего осмотра составил от 6 до 17 лет. Длительность наблюдения в сроке 3–5 лет – 9 пациентов, 6–12 лет – 6 пациентов. У 2 пациентов эпилептическая энцефалопатия проявлялась в форме синдрома Ландау-Клеффнера, у 3 – синдрома псевдоленнокса, у 10 – ESES. Использовались следующие методы: клинико-неврологический, интервьюирование родителей, анализ документации (заключения логопеда, психолога, характеристика из ДДУ, школы), ЭЭГ-видеомониторирование.
В активном периоде ЭЭ с CSWS эпилептические припадки наблюдались у 80% пациентов, когнитивные нарушения – у 73%, расстройства поведения – у 67%, нарушения речи – у 60%, неврологические нарушения – у 53%. Возраст появления основных клинических синдромов был следующим: нарушения речи – 2,5–5 лет, нарушения поведения – 3,5–5 лет, эпилептические припадки – 2,5–7 лет, когнитивные нарушения – 4–7 лет.
Среди синдромов ЭЭ с CSWS по степени выраженности наиболее мягким течением отличались эпилептические припадки (в 25% – редкие), наибольшей тяжестью – когнитивные нарушения (в 54% – выраженные). Анализ динамики клинических синдромов показал отчетливую тенденцию эпилептических припадков к снижению частоты и формированию ремиссий (50%), тогда как когнитивные нарушения в 27% наблюдений имели прогрессирующее течение.
86,7% пациентов получали терапию антиконвульсантами (40% – монотерпию, 46,7% – политерапию): вальпроат (депакин, конвулекс) – 100%, леветирацетам (кеппра) – 46%, топирамат – 31%, этосуксимид – 31%, ламотриджин – 15%. 80% получали лечение кортикостероидами.
Наступление резидуального периода ЭЭ с CSWS оценивалось по следующим критериям: спонтанный устойчивый (не менее 6 месяцев, при 2-кратном проведении ЭЭГ-мониторирования) регресс ЭЭГ-паттерна CSWS; прекращение эпилептических припадков; при синдроме ЛандауКлеффнера – устойчивый регресс афазии. Резидуального периода достигли 6 пациентов (3 мальчика, 3 девочки), из них в 2 наблюдениях с синдромом ЛандауКлеффнера, в 1 – с синдромом псевдоленнокса, в 3 – с ESES. Активный период заболевания длился у них от 1 до 8 лет, эпилептические припадки существовали в среднем 3 года. Резидуальный период наступил в возрасте 10,5–12 лет, продолжительность наблюдения в резидуальном периоде составила от 1 до 7 лет. Сравнение динамики клинических синдромов от активного к резидуальному периоду показало наибольшую динамичность эпилептического синдрома (регресс 100%), эпилептической активности (регресс 67%), тогда как когнитивные нарушения и расстройства поведения не претерпели динамики у всех пациентов.
Резюмируя сказанное, можно отметить следующее. Манифестными симптомами дебюта ЭЭ с CSWS в наших наблюдениях были эпилептические припадки или острые нарушения речи. Диагностика заболевания осуществлялась в минимальный срок. На протяжении активного периода наиболее благоприятной была динамика эпилептических припадков, неблагоприятной – когнитивных нарушений. Хотя возраст завершения активного периода в наших наблюдениях оказался несколько более ранним, чем по литературным данным, длительность активного периода была достаточно высокой, что ухудшало прогноз заболевания. К сожалению, доступные в настоящее время терапевтические воздействия на ЭЭ с CSWS недостаточно эффективны, что заставляет считать проблему ЭЭ с CSWS одной из наиболее актуальных и нерешенных в детской эпилептологии.
Делегаты конференции прослушали также интересную историческую информацию, предоставленную компанией Санофиавентис груп об оригинальном препарате Депакин.
Была представлена историческая справка, в которой сообщалось, что впервые вальпроевая кислота была зарегистрирована в виде оригинального препарата Депакин. В сообщении освещались вехи истории и была восстановлена хронология появления препаратов вальпроевой кислоты СССР и в мире. Было показано, что препарат Депакин был первым вальпроатом, который был зарегистрирован в СССР, правопреемником которого является Российская Федерация, это произошло в августе 1979г. Впервые о противоэпилептических свойствах вальпроевой кислоты заявили французские ученые H.Meunier, Y.Meunier, P.Eymard, P.Carraz. Исследования были проведены в Гренобле в 1962г. в исследовательском центре Laboratoire Berthier, был получен патент (FR2442M) на противоэпилептические свойства вальпроевой кислоты. Результатом исследований стала публикация научного открытия в журнале Therapie в 1963г. В дальнейшем лаборатория Berthier уступила право на разработку и распространение вальпроевой кислоты компании Labaz, впоследствии сформировавшей основу компании Санофи. В 1967г. вальпроевая кислота в виде препарата Депакин была впервые зарегистрирована в Европе, вначале во Франции, затем в Испании – в 1970г., Бельгии и Нидерландах – в 1971г., Великобритании, Швейцарии и Италии – в 1972г., ФРГ – в 1973г. и уже к первой половине 70-х годов Депакин получил широкое распространение в Европе. В августе 1979г. Депакин стал первым зарегистрированным вальпроатом в Советском Союзе.
Компания Санофи является первооткрывателем, разработчиком и производителем оригинального препарата Депакин и представленной в виде различных лекарственных форм, новейшей из которых является Депакин хроносфера, удовлетворяющий современным стандартам лечения эпилепсии.
И.Н. Вакула (к.м.н., главный детский психиатр Краснодарского края, Краснодарский государственный медицинский университет) представила доклад «Проблема диагностики и лечения аутизма» с соавторами Е.И. Пономаренко, Л.В. Царегородцевой, Е.Ю. Никифоровой.
Тесная связь признаков органического поражения головного мозга и поведенческих, когнитивных и аффективных расстройств у детей с аутизмом приводит к заключению, что они не просто сосуществуют и осложняют друг друга, но образуют этиопатогенетически целостное состояние (Каган В.Е.,1981). При детском аутизме целостная картина «составляется» только при тщательном анализе клинического, психологического и нейрофизиологического аспектов. Нам представляется, что последний недостаточно учитывается при обследовании детей с аутизмом, а это влияет на выбор фармакотерапии и, в конечном итоге, на его прогноз.
На 01.01.2012г. под наблюдением детских психиатров Краснодарского края находились 839 (655 мальчиков и 184 девочки) детей с аутизмом, синдромом аутизма, аутистическиподобными расстройствам, что составляет 9,5 на 10 тыс. детского населения. Соотношение девочек и мальчиков: 1: 3,6.
В связи с отсутствием «ответа» или наличием парадоксального или неадекватного «ответа» на антипсихотические препараты детям с аутизмом (с отсутствием эпилептических припадков) проводился ночной ЭЭГ – видеомониторинг на предмет наличия патологических паттернов. Из 83 обследованных была выявлена группа детей из 28 (34+5,2%) человек в возрасте от 3 до 13 лет (модальный возраст 6 лет), из них – 23 мальчика и 5 девочек с диагнозом: детский аутизм на резидуально-органическом фоне (25+8,2%), атипичный аутизм на резидуальноорганическом фоне (43+9,5%), атипичный аутизм с умственной отсталостью на резидуально-органическом фоне (32+9,5%). Метод ночного ЭЭГ-видеомониторинга позволил нам выявить качественные и количественные характеристики патологической активности и ее локализацию.
При сопоставлении патологических паттернов ЭЭГ: на первом этапе без учета клинической картины, на втором – путем сравнения качественных характеристик с целью выделения сходных по ЭЭГ- групп, на третьем – путем сопоставления выделенных групп с клинической картиной, выявилась корреляция типологической структуры ЭЭГ с психопатологическими симптомами. Структура эпилептиформных ЭЭГ-паттернов в исследуемых группах была неоднородной и представлена в виде: унилатеральных комплексов «острая-медленная волна» амплитудой от 90 до 600 мкВ, комплексов «множественный пик-медленная волна», часто в сочетании с региональными замедлениями и склонностью к вторичному диффузному распространению эпилептиформной активности; разрядов высокоамплитудных билатерально синхронных острых дельта волн с бифронтальным амплитудным преобладанием. Кроме того, отмечалось значимое преобладанием региональных патологических изменений БЭА мозга в лобно-височной и теменно-центральной областях правого полушария.
Таким образом, при сопоставлении групп по ЭЭГ с клиническими группами выявилась корреляция между степенью выраженности когнитивного дефицита и билатерально синхронной организацией и вторичным диффузным распространением эпилептиформной активности. Иными словами, вовлечение в эпилептический процесс неспецифических структур мозга разных уровней может вызывать нарушение работы различных модально-неспецифических систем. Могут возникать: нейродинамические нарушения высших психических функций в виде снижения их скорости, продуктивности, неравномерности; к динамической группе симптомов относится и изменение общего функционального состояния мозга, его колебания, истощаемость; избирательные нарушения эмоциональных процессов различной степени выраженности; нарушение цикла «сонбодрствование»; мнестические нарушения с первичными расстройствами кратковременной памяти: нарушаются механизмы произвольного запечатления и произвольного воспроизведения материала со снижением произвольного внимания.
На основании выявленной ЭЭГ-патологии детям с аутизмом к проводимому лечению или взамен принимаемых ими препаратов назначались антиконвульсанты. Наибольшее число детей (14 человек) получали вальпроаты (депакин, конвулекс) в дозе 30–60 мг/кг/сут. Хороший эффект был получен при применении леветирацетама (кеппры) у 7 пациентов в дозе 30–50 мг/кг/сут. Кроме того, назначался ламотриджин (ламиктал) 2 пациентам в дозе 5 мг/кг/сут., окскарбазепин (трилептал) – 3 пациентам в дозе 20–30 мг/кг/сут. и топирамат (топамакс) – 2 пациентам в дозе 5 и 6 мг/кг/сут.
По данным исследования мы пришли к некоторым выводам.
Ночной ЭЭГ-видеомониторинг является необходимым методом обследования для контингента детей с аутизмом как несущий важную диагностическую информацию. Количественные и качественные характеристики ЭЭГ коррелируют с тяжестью проявлений аутизма и нарушением когнитивных функций. При наличии эпилептической активности у детей с аутизмом введение в схему лечения антиконвульсантов, как правило, ускоряет положительную динамику клинической картины. Наилучший эффект был получен при применении вальпроатов и леветирацетама. На фоне лечения антиконвульсантами эпилептиформная активность регрессировала через 6–12 месяцев, но данный феномен не всегда коррелировал с соответствующей положительной клинической динамикой. Ни в одном случае не наблюдалось отрицательного влияния антиконвульсантов на когнитивные функции.

Докладчики и делегаты Конференции.
Д. м. н., профессор ГБОУ ВПО РНИМУ им. Н.И. Пирогова, вицепрезидент НП «Объединение врачей-эпилептологов и пациентов» К.В. Воронкова представила доклад «Эпилепсия и аутизм» совместно с О.А.Пылаевой и профессором, д. м. н. А.А. Холиным.
Эпилептические синдромы, наиболее часто ассоциирующиеся с аутизмом, включают синдром Веста, особенно при туберозном склерозе, синдром ЛандауКлеффнера, когнитивную дезинтеграцию с паттерном продолженной пик-волновой активности в медленном сне, атипичную роландическую эпилепсию, синдром Драве, синдром Леннокса-Гасто (Besag F.M., 2004) и другие.
Описана группа когнитивных дезинтеграций, в основе которых – паттерн продолженных пик-волновых разрядов во время сна (Зенков Л.Р., 2007; Ермоленко Н.А., 2009; Мухин К.Ю., 2011; Холин А.А., Воронкова К.В., 2012). У части пациентов клинические проявления эпилептических приступов отсутствуют, однако продолженная эпилептиформная активность во сне становится причиной когнитивной дисфункции – нарушений речи, памяти, способностей к обучению, поведенческих расстройств, в том числе, и аутистического поведения. Преобладание в клинической картине аутистических черт позволило выделить два отдельных синдрома в рамках этой группы эпилептических энцефалопатий. Термин «аутистический эпилептический регресс» применяется у детей с аутизмом, имеющих указание на регресс развития в анамнезе и эпилепсию. Термин «аутистический регресс с эпилептиформными изменениями на ЭЭГ» применяется у детей с аутизмом, не имеющих эпилептических приступов, у которых обнаруживаются эпилептиформные изменения на ЭЭГ (McVicar K.A., Shinnar S., 2004). Это состояние рассматривается, как вариант синдрома Ландау-Клеффнера. Предполагается, что оба заболевания могут иметь общий патогенез (Lewine J.D. и соавт., 1999).
Нами наблюдалось 113 детей, которые были разделены на группы: 1 группа (27 детей) – с органическим аутизмом; 2 группа (25 детей) – с наследственно-дегенеративными заболеваниями; 3 группа (11 детей) – с эпилептическим аутистическим регрессом; 4 группа (19 детей) – с аутистическим регрессом (с когнитивной дезинтеграцией) с эпилептиформной активностью на ЭЭГ; 5 группа (14 детей) – с сочетанием, ассоциированным с эпилептиформной активностью на ЭЭГ, но с невысоким индексом представленности; 6 группа (17 детей) – с процессуальным аутизмом. Во 2 и 3 группах преобладали девочки, в остальных – мальчики. В 1 и 2 группе нарушения дебютировали до года, в 3 и 4 – до 1,5 до 2 лет. В неврологическом статусе различные варианты очаговой неврологической симптоматики преобладали у пациентов 1 и 2 групп, что коррелировало с изменениями при проведении нейровизуализации. У пациентов 3, 4 и 5 групп на ЭЭГ наиболее часто регистрировались паттерны типа ДЭРД, индекс которых нарастал во сне, регистрировавшиеся преимущественно в височных, центральных и теменных отведениях.
У пациентов 1 и 2 групп – преобладало замедление фоновой активности, отмечалось региональное продолженное замедление, и эпилептиформная активность, паттерны типа ДЭРД отмечались у 8% и 6% больных соответственно. У пациентов 6 группы эпилептиформная активность не регистрировалась.
В целом наличие эпилептиформной активности в бодрствовании было выявлено у 63% больных, из них у 45% были выявлены ДЭРД. Выявлено существенное нарастание индекса эпиактивности во сне, по сравнению с бодрствованием – у 94% пациентов. Причем, у пациентов именно с эпиаутизмом (не симптоматическим) эпиактивность во сне выявлялась в 100% случаев. Кроме того, во сне выявлено наличие эпилептиформной активности еще у 14% больных человек с отсутствием таковой в бодрствовании.
Основной составляющей терапии у наблюдавшихся пациентов являлись вальпроаты (Депакин хроносфера, Депакин хроно и другие) которые назначались в 67% случаев: 32% – в монотерапии; 41% – в дуотерапии (с суксилепом, кеппрой, осполотом, топамаксом, финлепсином, гексамидином); 27% – три АЭП в комбинации (с суксилепом, кеппрой, топамаксом, ламикталом/ламолепом, финлепсином/тегретолом, фризиумом).
После оптимизации терапии пациенты получали: Депакин-хроносфера/хроно 20–50 мг/кг/сут. – 53%; Депакин-хроносфера/хроно+ кеппра 20–50 мг/кг/сут. – 30%; Депакин-хроносфера/хроно+суксилеп 10–30 мг/кг/сут. – 5%; кеппра+конвулекс – 10%; кеппра+ суксилеп – 5%; Депакин-хроносфера/хроно+осполот 4–6 мг/кг/ сут. – 2%.
У большинства пациентов отмечалась редукция эпиактивности на ЭЭГ, однако нарушения в высшей психической сфере сохранялись.
Кроме того, пациенты получали дополнительную терапию, включавшую: пантокальцин, кортексин, церебролизин, ноотропил, когитум, энцефабол, мексидол, танакан, актовегин, глицин, глиатилин, фенибут, магне В6, циннаризин, кавинтон, рибоксин, мильгамма, элеутерококк, элькар, новопассит, пустырник, сонапакс, терален, неулептил. Связь между эпилепсией и аутизмом до сих пор недостаточно изучена, однако с клинической точки зрения существование этой связи должно быть своевременно диагностировано, и обследование для исключения эпилепсии (ночной видео ЭЭГмониторинг) должно проводиться в рутинном порядке у пациентов с аутизмом (Canitano R., 2007).
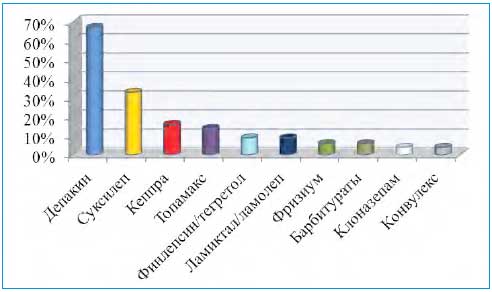
Терапевтическая тактика (собственное исследование) при лечении пациентов с аутистическими чертами и эпилепсией.
Большинство авторов назначает пациентам с эпиаутизмов комбинированную терапию, чаще вальпроата с сукцинимидами, осполотом или кеппрой. В условиях наличия/отсутствия в России некоторых АЭП, рациональной комбинацией является вальпроат+леветирацетам (кеппра). По мнению R.Guerrini, 2006, из вальпроатов именно Депакин хроносфера является препаратом выбора в лечении заболеваний с наличием аутистических черт и с моторными нарушениями.
Прозвучали практические доклады по диагностике эпилепсии. В видеопрезентации Оливье Дюлака (MD, Hospital Necker, Paris) была представлена «Диагностика эпилепсий у детей». Профессор С.К. Евтушенко (д.м.н., заведующий кафедрой детской неврологии факультета последипломного образования Донецкого Национального Медицинского Университета им. М. Горького, заслуженный деятель науки Украины) представил фундаментальный доклад «Катастрофические формы эпилепсии и эпилептические энцефалопатии у детей».
Н.Ю. Королева (Институт мозга человека им. Н.П.Бехтеревой РАН, СПб) представила случай из практики.
Е.С. Ильина (Российская детская клиническая больница, заведующая 2 психоневрологическим отделением, к.м.н.) сделала доклад «Практические аспекты терапии эпилептических энцефалопатий».
А.А. Холин (Российский национальный исследовательский медицинский университет им. Н.И.Пирогова, д.м.н., профессор) сделал доклад «Злокачественные мигрирующие парциальные приступы младенчества и синдром Марканда-Блюме-Отахара. Диагностика, терапия». Злокачественные мигрирующие парциальные приступы младенчества (ЗМППМ) дебютируют в возрасте до 6 месяцев жизни и характеризуются множественными и продолжительными электроклиническими фокальными приступными паттернами с вовлечением различных независимых отделов обеих гемисфер, регрессом и грубой задержкой психомоторного развития и неблагоприятным прогнозом для развития ребенка и приступов, в том числе и высокой летальностью в течение первого года жизни. В отделении ПНО-2 Российской Детской Клинической Больницы было зафиксировано 19 случаев детей с данной формой заболевания. У всех детей (100%) отмечались тонические спазмы, адверсивные тонические приступы, офтальмотонические приступы, а также у всех приступы протекали в виде эпилептического статуса мигрирующих фокальных приступов. У 7 детей диагностировалась «классическая форма», у 5 детей – «умеренный вариант», у 5 детей – «микст-форма» и у 2 детей – «стертая форма».
В качестве АЭП применялисьвальпроаты, бензодиазепины, этосуксимид, барбитураты, леветирацетам, руфинамид, бромиды и витамин В6 в высоких дозах. Препараты карбамазепинового ряда и вигабатрин противопоказаны при данном заболевании, в связи с высоким риском аггравации приступов. А в качестве купирования рефрактерного эпилептического статуса применялись в/в вальпроаты, оксибутират натрия, пропофол и тиопентал натрия.
Тяжелая эпилепсия с множественными независимыми фокусами спайков или синдром Марканда-Блюме-Отахара, Severe Epilepsy with Multifocal Independent Spike Foci (SE-MISF), характеризуется очень частыми и различного типами приступов, преимущественно малые моторные и короткие «псевдогенерализованные», паттерном MISF на ЭЭГ (множественные независимые очаговые спайки и пик-волновые комплексы, три и более очага в обеих гемисферах и не менее одного в каждой гемисфере) в сочетании с диффузным замедлением фоновой активности, отставанием в психическом, и, часто, также в моторном развитии, различной очаговой неврологической симптоматикой и неблагоприятным прогнозом, как для приступов, так и для психомоторного развития. Было проанализировано 12000 пациентов и из них у 443 (3,7%) был выявлен паттерн MISF. Среди популяции 443 детей с MISF обнаружено 117 случаев удовлетворявших вышеописанным критериям, что составило 26,4%. У всех детей в клинической картине присутствовали тонические спазмы (100%), а также доминировали миоклонические (62,4%), фокальные адверсивные (43,6%) и атипичные абсансы (36,8%). Этиология данного заболевания весьма разнообразна, но доминирующим фактором являлось гипоксически-ишемическое поражение ЦНС (40,2%). Также были отмечены травматические и инфекционные поражения ЦНС, пороки развития головного мозга, наследственные болезни обмена, хромосомные заболевания и др. Криптогенных форм отмечено 6%. В качестве препаратов выбора для лечения SE-MISF предпочтительнее вальпроаты, бензодиазепины, этосуксимид, леветирацетам и руфинамид. Также была проанализирована эволюция в данной выборке: для 71% –это конечная форма эпилептической энцефалопатии младенческого и раннего детского возраста, у 28% эволюционных изменений не отмечалось, и у 5% больных отмечалась трансформация в когнитивную эпилептиформную дезинтеграцию.
И.Д. Лемешко(Центральная клиническая больница Российской Академии Наук, к.м.н.) привела «Случай из практики. Инфантильные спазмы. Поздняя форма». Эпилептические спазмы, как считалось ранее, характерны для детей 1-го года жизни и зачастую рассматривались в рамках синдрома Веста. Однако в последние годы было проведено несколько независимых исследований, в которых были выявлены дети с дебютом спазмов после 12 месяцев жизни. В данном докладе был приведен клинический пример эпилептической энцефалопатии с поздним дебютом спазмами у мальчика, 4лет. До дебюта заболевания ребенок рос и развивался нормально, занимался танцами, читал стихи, «был отрадой для всей семьи». В 2,5 года появились приступы в виде замираний и отведения глаз вверх, с частотой до нескольких раз в день. Через 1 месяц присоединились аксиальные тонические спазмы с «кивками» и генерализованные тонико-клонические судорожные приступы. Мальчик стал отставать в психоречевом развитии. Был назначен Депакин хроно 300 мг/сут., без эффекта и в течение последующего года обследовался и лечился в клинике Израиля с диагнозом синдром Леннокса-Гасто, где ему подбиралась антиэпилептическая терапия: вальпроевая кислота + клобазам – без эффекта, вальпроевая кислота + топирамат + клоназепам – выраженный седативный эффект и вялость, вальпроевая кислота + зонисамид – без эффекта. К 3,5 годам по возвращению в Москву ребенок утратил речевые навыки, общался жестами, говорил только несколько слов. Приступы участились до 30 приступов в серию и до 15 серий в сутки. При осмотре в неврологическом статусе очаговой симптоматики не выявлено. На МРТ головного мозга – данных за очаговые изменения в веществе головного мозга нет. В октябре 2011г. был проведен ночной видео-ЭЭГ мониторинг (на фоне приема вальпроевой кислоты, клобазама и леветирацитама): была зарегистрирована выраженная мультифокальная и диффузная эпилептиформная активность, исходящая преимущественно из лобной доли и зафиксированы приступы по типу тонических аксиальных спазмов, сопровождающиеся на ЭЭГ диффузными пробежками быстрых форм бета-диапазона в сочетании с короткими периодами уплощения б.э.а., а также многочисленные субклинические иктальные ЭЭГ-паттерны спазмов. Ребенок был проконсультирован профессором О.Дюлаком, который диагностировал у него «Эпилептическую энцефалопатию с поздним дебютом инфантильных спазмов» и назначил гидрокортизон в монотерапии по схеме в высоких дозах с отменой других АЭП. На 1-ой неделе приема гидрокортизона (150 мг/сут) развился тяжелый приступ аксиальных спазмов со статусным течением. В ноябре к гидрокортизону (80 мг/сут) был добавлен топирамат 62,5 мг/сут, у ребенка прекратилась экспрессивная речь. При проведении ЭЭГ отмечена положительная динамика в виде снижения амплитуды и индекса эпиразрядов, увеличения «светлых эпох», но во сне – увеличилось число иктальных паттернов аксиальных спазмов и офтальмотонических приступов и присоединение нового типа приступов – миоклонических. Последующее снижение дозы топирамата до 50мг/сут привело к улучшению речевой продукции. Со слов родителей на фоне приема гидрокортизона в течение 2 месяцев кинематика и частота приступов не менялась. Ребенок сильно прибавил в весе (+7–8кг), появился гипертрихоз лица и туловища. Он стал более агрессивным, капризным, отмечалось повышение апетита (при отказе в еде грыз стол). Со слов мамы «он не думал о еде только в процессе кормления, поэтому приходилось его постоянно чем-нибудь кормить маленькими порциями». В феврале 2012г. на фоне монотерапии гидрокортизоном на ЭЭГ в бодрствовании появилось больше «светлых эпох» на ЭЭГ, наросли частотные характеристики физиологических ритмов, а во сне не было снижение индекса и амплитуды эпилептиформных разрядов. Но, учитывая недостаточную эффективность гормональной терапии, было принято решение о ее отмене и введение депакин хроносфера 750мг/сут + кеппра 1000мг/сут, на которой он находится до настоящего времени. Впервые данная форма эпилептической энцефалопатии была описана во Франции в середине ХХ века – 18 случаев. Она, также как и синдром Веста, подразделяется на криптогенную и симптоматическую. Возраст дебюта варьирует от 12 месяцев до 12 лет жизни. Заболевание может начинаться не только со спазмов, но также с любого вида приступов. Приступы серийные, чаще асимметричные спазмы, а также возможно сочетание спазмов с миоклоническими, парциальными приступами или абсансами. Изменения в неврологическом статусе чаще отмечались у детей с симптоматической формой. После дебюта заболевания в большинстве случаев отмечается регресс психоречевого развития, а также ЗПРР может быть изначально. На интериктальной ЭЭГ у детей с данным синдромом типичная гипсаритмия встречается около у 15% больных. У большинства наблюдается высокоамплитудная мультифокальная спайк-полиспайк-волновая активность, диффузное замедление б.э.а., персистирование в фазу медленного сна и исчезновение в фазу REM-сна. По данным многих авторов паттерны имеют лобную и височную акцентуацию. Данная форма фармакорезистентна к базовым и новым препаратам. По некоторым данным положительный эффект был зафиксирован на фоне приема вигабатрина и гидрокортизона. Эволюция возможно в синдром Леннокса-Гасто или отмечается персистирование приступов в течение всей жизни с резистентностью к любой антиэпилептической терапии. Также описано несколько случаев полной клинико-ЭЭГ ремиссии.

Участники конференции участуют в дискуссии. Главный детский эпилептолог г. Казань Е.А. Морозова.
По окончании заседаний состоялась продолжительная и интересная дискуссия. Присутствовавшие на Конференции участники единогласно одобрили инициативу проведения подобного мероприятия в рамках празднования Европейского Дня эпилепсии в России.
Материал подготовлен К.В. Воронковой. В статье размещены авторские резюме докладов, присланные в редакцию: К.Ю. Мухиным, Н.А. Ермоленко, Н.Ю. Перуновой & Т.Р. Томенко, И.Н. Вакулой, А.А. Холиным, И.Д. Лемешко. Фото Г.В. Катиной-Ярцевой.
