Роль гликирования белков, окислительного стресса в патогенезе сосудистых осложнений при сахарном диабете
СтатьиМ.И. Балаболкин
ГУ Эндокринологический научный центр (дир. ~ акад. РАМН И. И. Дедов) РАМН, Москва
Сахарный диабет (СД) является фактором риска развития ангиопатий (микро- и макроан-гиопатии), которые являются причиной высокой инвалидизации и летальности. В соответствии с современными данными в патогенезе сосудистых осложнений при СД типа 2, помимо гипергликемии, недостаточности функции р-клеток, инсулиновой резистентности и гиперинсулинемии, участвуют дополнительные группы факторов риска (рис. 1).
Дислипидемия при СД характеризуется повышением содержания общего холестерина, холестерина липопротеидов низкой плотности и триглицеридов липопротеидов очень низкой плотности, снижением холестерина липопротеидов высокой плотности.
Нарушения коагуляции проявляются снижением фибринолиза, повышением содержания фибриногена, а также увеличением экспрессии активатора тканевого плазминогена и ингибитора 1 типа активатора плазминогена (ИАП-1 или PAI-1).
Воспаление, которое постоянно встречается при многих хронических системных заболеваниях, включая сахарный диабет, проявляется повышением экспрессии его специфических маркеров: фибриногена, С-реактивного и белка амилоид -1, а также повышением уровня цитокинов (интерлейкины, ос-фактор некроза опухолей и др.).
Окислительный стресс, являющийся следствием повышенного аугоокисления глюкозы, проявляется повышением продуктов перекисного окисления липидов, окисленных липопротеидов низкой плотности и производных арахидоновой кислоты (Р2-изопростаны).
Повышение гликирования белков сопровождается увеличением продуктов конечного гликозилирования, которые инициируют экспрессию генов коллагена и других белков капиллярной мембраны, обладающих проатерогенными свойствами.
Дисфункция эндотелия сопровождается повышением экспрессии генов клеточных адгезивных молекул и ИАП-1, а также снижением образования оксида азота, обладающего вазодилатирующим действием. Рассматривая сложный «многоступенчатый и многокомпонентный» процесс, приводящий к развитию ангиопатий, целесообразно вначале осветить роль гликирования белков и окислительного стресса, которые инициируют дисфункцию эндотелия, и другие компоненты, участвующие в патогенезе сосудистых осложнений диабета.
Рис. 1.
Патеногенез сосудистых осложнений СД типа 2.
|
Сахарный диабет типа 2: | ||||||
|
↓ |
↓ |
↓ |
↓ |
↓ |
↓ |
↓ |
|
Дисфункция эндотелия |
Окислительный стресс |
Полиоловый путь |
Нарушение коагуляции |
Гликирование белков |
Воспаление |
Дислипидемия |
|
↓ |
↓ |
↓ |
↓ |
↓ |
↓ |
↓ |
|
Сосудистые осложнения диабета | ||||||
Гликирование, или гликозилирование, обусловлено способностью глюкозы образовывать с аминогруппами различных белков, возможно и с ДНК, различные соединения (интермедиаты), участвующие в обмене и являющиеся исходным материалом для образования необратимых в химических реакциях веществ, которые получили название конечных продуктов гликозилирования (КПГ). Период полураспада этих продуктов более длительный, чем белков (от нескольких месяцев до нескольких лет). Скорость образования КПГ зависит от уровня и длительности экспозиции глюкозы.
В зависимости от уровня гликемии в крови КПГ образуются сравнительно быстро как внутри-, так и внеклеточно. У диабетических крыс КПГ (концентрация последних определялась с помощью специфической флюоресценции) увеличивались в сосудах сетчатки в 2,6 раза уже через 26 нед. от начала экспериментального диабета. Подобное накопление конечных продуктов гликозилирования наблюдается в белках хрусталика и в коре почек диабетических животных. Усовершенствование методики определения количественного накопления КПГ (специфическое связывание КПГ соответствующими специфическими антителами) позволило установить, что в тканях диабетических животных уже через 5-20 нед. от начала диабета количество КПГ, связанных с антителами, увеличивается в 10-45 раз по сравнению с недиабетическими животными. Более того, основное количество КПГ в тканях диабетических животных приходится на КПГ, связываемых антителами, а не на количество КПГ, определяемых с помощью специфической флюоресценции. Количество КПГ прямо пропорционально уровню глюкозы в крови, и даже умеренное повышение гликемии (7-8 ммоль/л) приводит к достоверному их увеличению.
В последние годы показано значение КПГ в механизмах развития сосудистых осложнений у больных СД. Образование иммунохимических КПГ предшествует ранним клиническим признакам ретинопатии и нефропатии у больных, страдающих инсулинзависимым сахарным диабетом (ИЗД). Так, в коже больных СД типа 1 задолго до появления самых ранних клинических признаков ретинопатии и нефропатии наблюдается увеличение количества КПГ. Более того, установлено, что при диабете иммунохимические (иммунореактивные) КПГ накапливаются в узловых и диффузных повреждениях клубочкового аппарата почек, а также в отложениях гиалина, локализованного в артериолах. Исследования с помощью сканирующей электронной микроскопии показали, что в клубочковом аппарате почек при наличии иммунохимических КПГ наблюдается увеличение размера пор матричного сита базальной мембраны, что объясняет повышение клубочковой проницаемости, наблюдаемой у больных СД. Аналогичное накопление иммунореактивных КПГ определяется в аорте и особенно в атеросклеротических бляшках.
Рис. 2.
Схема гликозилирования белков
|
Глюкоза, фруктоза, | |||||||
|
+ | |||||||
|
Эпсилон-аминогруппа (NH2) белка | |||||||
|
↑ |
↓ |
Минуты | |||||
|
Шифф-основание | |||||||
|
Лабильная форма | |||||||
|
Перестройка | ↑↓ |
Часы, дни | |||||
|
| |||||||
|
Стабильная форма | |||||||
|
↑ |
↓ |
Тиоктовая кислота | |||||
|
Реактивные карбониловые интермедиаты: 3-деоксиглюказон, метилглиоксоль | |||||||
|
| |||||||
|
Неактивные метаболиты |
Конечные продукты гликозилирования | ||||||
|
↓ | |||||||
|
Индукция экспрессии различных генов и активация транскрипционных факторов, включая NF-kB | |||||||
Гликозилирование белков и образование КПГ — сложная многоэтапная цепь метаболических процессов. Первым этапом гликозилирования (рис. 2) является образование альмидина (N-гликозиламина) или соединения глюкоза-белок. Альмидин является лабильным и обратимым соединением, для образования которого требуется всего несколько часов. При условии сохранения повышенного уровня глюкозы образуется вещество Амадори (1-амино, 1-деоксикетоза), стабильная форма (рис. 3), которая окисляется в так называемые «реактивные дикарбониловые интермедиаты» (3-деоксиглюкозон и метил глиоксаль). Окисляясь дикарбоншювые интермедиаты превращаются в КПГ (рис. 4). Кроме того, специфические редуктазы процессом «детоксикации» могут трансформировать дикарбониловые интермедиаты в неактивные метаболиты. Второй путь метаболизма дикарбониловых интермедиатов предпочтителен, так как его конечные продукты не участвуют в механизмах повреждения функции многих белков и тканей. Из данных на рис. 3 и 4 видно, что тиоктовая, или ос-липоевая, кислота снижает количество реактивных карбониловых интермедиатов, улучшает активность редуктазы, что приводит к уменьшению образования конечных продуктов гликозилирования, следовательно, к снижению инициации и скорости прогрессирования сосудистых осложнений диабета.
В последние годы показано, что КПГ могут образовываться другим более коротким метаболическим путем, т.е.путем металкатализуемого аутоокисления различных Сахаров и образования их них реактивных дикарбониловых интермедиатов, минуя перечисленные этапы метаболизма. Так, в исследованиях in vitro установлено, что около 50% КПГ (в частности, карбоксиметиллизин) образуются путем окисления вещества Амадори, а около 50% — другими метаболическими путями, включая аутоокисление различных Сахаров.
До последнего времени глюкозу считали основным веществом для образования КПГ. Установление различной скорости внутри- и внеклеточного образования КПГ позволило усомниться в этом. Оказалось, что скорость более быстрого внутриклеточного образования КПГ определяется такими сахарами, как фруктоза, глюкозо-6-фосфат и глицералальдегид-3-фосфат. Кроме того, различными иммунохимическими исследованиями с помощью аутоантител установлено, что в составе КПГ содержатся различные сахара.
Эритроциты генетически ожирелых и стрепто-зотоциновых мышей содержат глиоксалазную активность, необходимую для образования 3-деоксиглюкозона, 3-деоксифруктозы и метилглиоксаля, уровень которых повышен при СД. Глиоксалазная система, контролирующая образование КПГ в различных органах и тканях, практически независимо от уровня глюкозы в крови является одним из важнейших регуляторов биологических процессов в организме, определяющих нормальное функционирование организма и длительность его жизни. Глиоксилаза-1 необходима для превращения метилглиоксаля сначала в S-D-лактоилглютатион, а затем в D-лактат. В 1993 г. был идентифицирован и клонирован основной 3-деоксиглюкозонредуцирующий фермент (2-оксо-альдегидредуктаза), который оказался идентичен альдегидредуктазе и который конвертирует 3-деоксиглюкозон в 3-деоксифруктозу. Таким образом, 2-оксоальдегидредуктаза или альдегидредуктаза является одним из ферментов, участвующих в детоксификации различных интермедиатов КПГ и, в частности, 3-деоксиглюкозона. Различные мутации и другие нарушения гена, ответственного за синтез 2-оксоальдегидредуктазы, будут сочетаться с высоким риском развития сосудистых осложнений диабета. Таким образом, один из реактивных интермедиатов КПГ -3-деоксиглюказон является продуктом как гликирования белков, так и полиолового пути обмена глюкозы (рис. 5).
Рис. 5.
Схема образования и накопления 3-деоксиглюказона (реактивный карбониловый интермедиат).
|
Глюкоза + белок |
→ |
Шифф-основание |
→ |
Вещество Амадори |
→ |
Фрукто-3-фосфат |
|
↑ | ||||||
|
Глюкоза |
→ |
Сорбитол |
→ |
Фруктоза |
→ |
Фрукто-3-фосфат |
* Альдозоредуктаза;
** сорбитолдегидрогеназа;
*** киназа
КПГ, в свою очередь, участвуют в механизмах развития поздних осложнений диабета несколькими путями. Быстрое образование внутриклеточных КПГ способствует нарушению функции внутриклеточных белков, и их количество в гемоглобине эритроцитов может служить объективным маркером конечного гликозилирования в тканях. У практически здоровых лиц содержание КПГ в эритроцитах составляет 0,42%, тогда как у больных СД — 0,75%. КПГ сравнительно быстро накапливаются в эндотелиальных клетках, где они выявляются в комплексе с фактором роста фибробластов, одним из ростовых факторов, принимающих участие в клеточном цикле и функции клеток. Кроме того, внутриклеточное увеличение КПГ снижает каталитическую активность альдегидредуктазы (2-оксоальдегидредуктазы), что ускоряет последующее дополнительное образование КПГ из реактивных дикарбоншювых метаболитов.
Внеклеточное накопление КПГ изменяет структуру и функциональные свойства как матрикса, так и матриксклеточных взаимодействий. КПГ ковалентно взаимодействуют с коллагеном I типа, который взаимодействует с такими различными растворимыми белками плазмы, как липопротеины низкой плотности, иммуноглобулин G и др. Образование КПГ на белках базальной мембраны (коллаген IV типа, ламинин, гепарансульфат протеогликан и др.) приводит к ее утолщению, сужению просвета капилляров и нарушению их функции (снижение адгезии эндотелиальных клеток, снижение пролиферации ретинальных перицитов, повышение пролиферации ретинальных эндотелиальных клеток и др.). Эти нарушения внеклеточного матрикса изменяют структуру и функцию сосудов (снижение эластичности сосудистой стенки, изменение ответа на сосудорасширяющее действие оксида азота и др.), способствуют более ускоренному развитию атеросклеретического процесса.
КПГ принимают непосредственное участие в экспрессии генов, ответственных за образование различных белков, принимающих участие в развитии патологических и морфологических структур. В экспериментальных исследованиях показано, что введение КПГ мышам в течение 4 нед. сопровождается увеличением экспрессии генов и повышением синтеза белков, что имеет прямую корреляционную зависимость с количеством соответствующих мРНК, в том числе гломерулярного а-1 коллагена IV типа, а также ламинина Bj и (3)-трансформирующего фактора роста.
Расшифрован механизм экспрессии генов под влиянием КПГ. Вначале указанные соединения связываются со специфическими КПГ-рецепторами, локализованными на моноцитах, макрофагах, эндотелиальных и других клетках, которые опосредуют трансдукцию этого сигнала посредством увеличения образования свободных радикалов кислорода. Последние в свою очередь активируют транскрипцию ядерного NF-kB фактора-регулятора экспрессии многих генов, отвечающих на различные повреждения. Это специфическое активирующее экспрессию различных белков действие КПГ может быть прервано или заблокировано применением антител к рецепторам КПГ или антител к КПГ.
В исследованиях на культурах клеток продемонстрировано, что при высоких концентрациях глюкоза с помощью фермента альдозоредуктазы превращается в сорбитол (рис. 5). Последний при участии фермента сорбитолдегидрогеназы мета-болизируется во фруктозу с образованием NADH. Скорость конверсии сорбитола во фруктозу значительно ниже, чем скорость образования последнего из глюкозы. Кроме того, сорбитол не диффундирует через мембрану клетки, что и является причиной повышения внутриклеточной его концентрации. Накопление сорбитола в клетке приводит к осмотическому стрессу. Именно этим объясняется нарушение зрения, часто наблюдаемое у больных сахарным диабетом при манифестации заболевания вскоре после начала инсулинотерапии. Нарушение углеводного обмена и гипергликемия, которая развивается до клинических проявлений сахарного диабета, приводят к увеличению обмена глюкозы по полиоловому пути и накоплению сорбитола и осмотическому отеку хрусталика глаза. Нарушается способность хрусталика к аккомодации. Однако нарушения зрения не наблюдается, так как мышцы глаза полностью компенсируют такую недостаточность хрусталика, которая развивается в течение длительного времени. Манифестация сахарного диабета и применение инсулинотерапии сравнительно быстро приводят к компенсации сахарного диабета и, естественно, к ингибированию полиолового пути обмена глюкозы и снижению концентрации сорбитола в хрусталике, т.е. к восстановлению функции хрусталика. Однако восстановление функции глазных мышц запаздывает, что и проявляется ухудшением зрения. Учитывая, что такие нарушения совпадают с началом инсулинотерапии, больные расценивают этот факт как побочное действие вводимого инсулина. Указанные явления со стороны зрения проходят самостоятельно через несколько дней.. Перед началом инсулинотерапии больных необходимо предупредить о возможном ухудшении зрения и что это не связано с побочным действием инсулина и не требует лечения. Тем не менее, повышение содержания сорбитола в хрусталике при длительной декомпенсации диабета может способствовать развитию катаракты, которая у больных диабетом развивается в более молодом возрасте по сравнению с лицами без диабета.
Исследования на животных с экспериментальным диабетом подтверждают значение повышенного образования сорбитола в патогенезе осложнений диабета. Нарушение функции периферических нервов и повышение скорости клубочковой фильтрации, которая наблюдается у животных с различными моделями диабета, нормализуется после назначения таким животным сорбинила — ингибитора альдозоредуктазы. Однако применение ингибиторов альдозоредуктазы для лечения поздних осложнений диабета не сопровождается столь значительными положительными эффектами, которые выявляются у диабетических животных.
В 1987 г. S. Wolff с сотрудниками одними из первых показали, что основная роль в развитии сосудистых осложнений диабета принадлежит неферментативному аутоокислительному гликозилированию и окислительному стрессу, вызванному нарушением углеводного обмена. Указанные нарушения способствуют усилению процессов пероксидации липидов и изменению качественных характеристик липопротеидов с их накоплением в пенистых клетках, являющихся основой атеросклеротического поражения крупных сосудов. Модифицированные липопротеиды также принимают участие в повреждении эндотелиальных клеток, способствуя развитию микроангиопатии. К сожалению, до настоящего времени отсутствуют методы, позволяющие непосредственно определять уровень окислительного стресса в организме. В этой связи состояние окислительного стресса и вызванные им нарушения и различные повреждения белков организма определяются косвенно по содержанию различных гликоокисленных продуктов, к которым относятся белковые карбонилы, липидные пероксиды и различные вещества, реагирующие с тиобарбитуровой кислотой. Свободнорадикальное окисление липидов является неотъемлемой частью таких жизненно важных процессов, как перенос электрона флавиновыми элементами, обновление состава липидов биомембран, окислительное фосфорилирование в митохондриях, митогенез, проведение нервного импульса и др. Продуктами перекисного окисления липидов (ПОЛ) являются предшественники простагландинов и их производных- тромбоксанов и простациклина. Постоянно протекающие в клеточных мембранах реакции пероксидации, способствуют обновлению их липидного состава и поддержанию соответствующей активности всех липидзависимых мембрано-связанных ферментов, к которым относятся практически все ферментные системы организма. Процессы ПОЛ представляют собой цепную реакцию и включают инициирование, удлинение, разветвление, обрыв цепей окисления. Основная роль в инициировании перекисных реакций принадлежит активным формам кислорода, таким как супероксидрадикал, синглетный кислород, гидроксил-радикал. Избыточное образование продуктов ПОЛ оказывает повреждающее действие на уровне клеток, и их цитотоксичность связана с накоплением перекисей липидов в липопротеидах высокой плотности (ЛПВП). При этом свободные радикалы участвуют в деструкции многих клеток, включая эндотелий.
Как известно, окислительные процессы в организме связаны с использованием кислорода по двум путям: 1) оксидазному, или основному, сопряженному с образованием АТФ, который и является главным источником энергии; 2) оксигеназному, характеризующемуся включением кислорода в молекулу окисляемого субстрата. При втором пути отсутствует полное восстановление кислорода до воды и образуются активные формы кислорода, такие как супероксидный анион, перекись водорода гидроксильный радикал. Последние активно реагируют с фосфолипидами и прежде всего с арахидоновой и докозогексаеновой кислотами мембран с образованием продуктов перекисного окисления. При распаде образовавшихся гидроперекисей появляется избыток свободных радикалов (RO*), несущих неспаренный электрон. Соединяясь с молекулой кислорода, они образуют новый радикал (RO2), который и называется перекисным.
Перекисные радикалы затем вступают во взаимодействие с молекулами жирных кислот, образуя высокотоксичные гидроперекиси (ROOH) и новый свободный радикал. Этот процесс протекает лавинообразно с увеличением концентрации свободных радикалов, которые затем снова формируют цепи окисления. Эта практически цепная реакция прерывается лишь взаимодействием с антиоксидантами.
Обладая высокой реактогенной способностью, свободные радикалы вступают в реакции с ненасыщенными жирными кислотами, являющимися компонентом мембранных фосфолипидов, в результате чего возникают новые цепи окисления, а в зонах наибольшей активности липопероксидации возникают каналы пассивной проницаемости, через которые свободно проходят ионы и вода. Диеновые конъюгаты, являющиеся первичными продуктами ПОЛ, относятся к токсическим метаболитам, которые оказывают повреждающее действие на липопротеиды, белки, ферменты и нуклеиновые кислоты. Дальнейшими продуктами ПОЛ являются альдегиды и кетоны (малоновый диальдегид и др.), которым принадлежит важная роль в синтезе простагландинов, прогестерона и других стероидов. Взаимодействие диальдегидов со свободными группами мембранных соединений образуют конечные продукты ПОЛ (основание Шиффа и др.), непрерывное накопление которых дестабилизирует мембраны и способствует деструкции клеток.
Дальнейшее увеличение количества свободных радикалов и гидроперекисей липидов должно было бы привести к быстрому разрушению клеточных структур, но в естественных условиях этого не происходит благодаря наличию сложной и многокомпонентной системы биоантиокислителей и естественных антиоксидантов, способных при химическом воздействии ингибировать свободнорадикальное окисление липидов. При нормальных условиях в организме сохраняется равновесие между скоростью ПОЛ и активностью антиоксидантной системы (витамины Е, С, В, супероксиддисмутаза, каталаза, глютатионтрансфераза, глютатионпероксидаза, глютатионредуктаза и др.), что является одним из основных показателей гомеостаза.
Избыточное образование продуктов ПОЛ, как отмечено выше, оказывает цитотоксическое действие, что проявляется повреждением мембран эритроцитов, лизосом. При этом изменяется структура мембран клеток, вплоть до их разрыва, ингибируется активность цитохромоксидазы.
Считается, что свободные радикалы участвуют в патогенезе многих заболеваний (по данных некоторых авторов этот перечень включает 100 различных болезней), в том числе ревматоидного артрита, геморрагического шока, язвенного колита, атеросклероза, синдрома приобретенного иммунодефицита и др. (рис. 6). Более того, свободные радикалы участвуют прямо или опосредованно в механизмах некроза и апоптоза, т.е. в процессах, регулирующих длительность тканей, органов и систем организма, в процессах старения организма и в конечном итоге, в контроле длительности жизни организма. Можно без преувеличения считать, что формируется новая область науки - свободнорадикальная биология и медицина.
Рис. 6.
Роль окислительного стресса при различных заболеваниях.
|
Свободные радикалы | ||||||||||
|
↓ |
↓ |
↓ |
↓ |
↓ |
↓ |
↓ |
↓ |
↓ |
↓ |
↓ |
| Сахарный диабет | Ката-ракта | Старение | Реперфузия после ишемии | Атеро-склероз | Болезнь Паркинсона | Болезнь Альцгеймера | Алкогольное/ лекарственное поражение печени | Ауто-иммунные заболе-вания | Рак | Артриты |
Многие свободные радикалы являются цитотоксическими и приводят к развитию патологических состояний. Однако свободнорадикальные реакции необходимы для образования многих жизненнонеобходимых ферментов, а также для нормальной функции иммунной системы и ее компонентов (нейтрофилы, макрофаги и другие иммунокомпетентные клетки), т.е. для формирования адекватного иммунного ответа. Установлено, что при воспалении фагоцитами и Т-лимфоцитами (Т-киллерами) активно образуются такие свободнорадикальные соединения, как О2*-, НОСl и NO*, которые обладают выраженным бактерицидным и противоопухолевым действием. Этот цитотоксический эффект свободнорадикальных соединений, который в норме выполняет защитную функцию (ликвидация патогенных микроорганизмов и мутантных клеток), при определенных состояниях (например, избыточное образование свободных радикалов, дефекты антиоксидантной системы) вследствие неконтролируемой их «утечки» может приводить к необратимым повреждениям молекул белков, липидов и нуклеиновых кислот.
Свободные радикалы также облигатны (абсолютно необходимы) для активации многих транскриптационных факторов, участвующих в экспрессии генов, а также осуществляют трансдукцию гормональных и клеточных сигналов. Многие свободные радикалы проявляют цитотоксические свойства в том случае, если они образуются в избытке, повреждая при этом как одиночные молекулы, так и мембраны клеток, клетки, ткани и органы. Развивается окислительный стресс и последующее лавинообразное образование свободных радикалов, приводящих к дальнейшей деструкции на клеточном и органном уровнях.
При физиологических условиях в организме имеется постоянный баланс между уровнем свободных радикалов (оксидантов) и активностью системы антиоксидантной защиты. Окислительный стресс сопровождается нарушением равновесия между указанными системами с увеличением количества оксидантов, которые ведут к повреждению биологических молекул в клетках, что сопровождается повышением их содержания в биологических средах и тканях организма. Соединения или вещества, представляющие собой такие поврежденные биологические молекулы (белки, липиды и др.), являются маркерами окислительного стресса.
Свободные радикалы представляют собой гетерогенную группу, но наибольшее их количество относится к соединениям реактивного кислорода, которые представляют собой как кислородные радикалы, так и нерадикальные соединения. Все представители этих соединений являются окислительными и легко конвертируются в радикалы.
Молекулярный кислород является наиболее распространенным и наиболее важным электронным акцептором. Обычно в митохондриях кислород восстанавливается 4 электронами до воды. При физиологических условиях лишь 5-6% потребляемого кислорода восстанавливается до супероксидных радикалов. При определенных условиях супероксидный радикал может восстанавливаться до гидроксильного радикала, который хотя и считается более слабым окислительным агентом, но в биологических системах обладает даже более выраженной реакционной способностью по сравнению с О2.
Все свободные радикалы имеют на внешней орбите неспаренный электрон, а для устойчивого состояния каждая молекула должна содержать на наружной орбитали два электрона. Поэтому второй недостающий электрон свободные радикалы захватывают из других молекул. Именно этим объясняется повышенная реакционная способность свободных радикалов. Супероксидные радикалы активируют перекисное окисление липидов. При взаимодействии с полиненасыщенными жирными кислотами супероксидный радикал в результате цепной реакции вызывает разрушение мембранных структур.
Таким образом, определенное количество свободных радикалов образуется постоянно в физиологических условиях. Однако при определенных условиях образование реактивных радикальных соединений увеличивается, что и приводит к окислительному стрессу. Причины, приводящие к окислительному стрессу, многочисленны. Это различные повреждения (травмы и др.), инфекции, избыточное ультрафиолетовое облучение, повышение уровня экзогенных и эндогенных токсинов, перегревание и резкое охлаждение организма, ишемическая реперфузия и др. Перечисленные состояния сопровождаются активированием различных ферментных систем (циклооксигеназы, конверсия ксантиндегидрогеназы в оксидазу и др.), высвобождением гембелков (миоглобин, гемоглобин, цитохромы ), которые реагируют с пероксидами и стимулируют свободнорадикальные повреждения различных клеток. Повреждения митохондрий сопровождаются повышенной утечкой электронов и образованием О2-, Н2О2. Повышается внутриклеточный уровень ионизированного кальция, что в свою очередь стимулирует Са2+-зависимые нуклеазы и Са2+-кальмодулинзависимую NO-синтазу с повышением образования NO, что создает возможность ускоренного образования ONOO. Активирование, таким образом, всех перечисленных процессов способствует развитию окислительного стресса.
Супероксидные радикалы активируют ПОЛ. Фо-сфолипиды, которые являются основными соединениями клеточной мембраны, вследствие их высокой ненасыщенности легко подвергаются повреждающему действию свободных радикалов. При их взаимодействии с полиненасыщенными жирными кислотами (свободные радикалы: супероксидный, гидроксильный и гидропероксидный) в результате цепной реакции происходит разрушение мембранных структур.
В процессе ПОЛ образуются вторичные соединения (липидные гидропероксиды, диеновые альдегиды — соединения альдегидной природы, к которым относится малоновый диальдегид и 4-гидроксиноненаль). Альдегидные группы этих соединений вступают в реакцию с аминогруппами белков и нуклеотидов, что приводит к нарушению структуры и функции таких молекул.
Окислительный стресс, сопровождающийся значительным увеличением уровня свободных радикалов и приводящий к повышению ПОЛ, блокирует синтез белка и нуклеиновых кислот, подавляет гликолиз и способствует разобщению окислительного фосфорилирования, ингибирует активность некоторых ферментов (глюкозо-6-фосфатазы, аденилатциклазы и др.), что приводит к нарушению функции многих тканей. Указанные изменения могут возникать в организме в тех случаях, когда скорость образования свободных радикалов превышает нейтрализующую способность ферментов антиоксидантной системы организма.
Антиоксидантная система организма представлена несколькими группами соединений: 1) ферменты: супероксиддисмутаза, глютатионпероксидаза, каталаза, селензависимый глютатион, тиоредоксин гидропероксидаза, метионинсульфоксид редуктаза, пероксиддисмутаза; 2) фитонутриенты: витамин С, витамин А, семейство токоферола (4 соединения токоферола — альфа- бета- гама- и дельта-токоферолы и 4 соединений токотриенолов), каротены (около 500), соединений флавониды и полифенолы (около 4000-5000 соединений), селений и различные микроэлементы, тиоктовая или липоевая кислота и ее восстановленная форма липоат (в восстановлении липоевой кислоты участвует несколько ферментов — глютатионредуктаза, тиоредоксин редуктаза и липамиддегидрогеназа); 3) секвестранты металлов: альбумин, трансферрин, ферритин, гемопексин; 4) другие антиоксиданты: билирубин, глютатион, таурин, убихинон (убиквинол, убифенол), ураты. Важным является тот факт, а-липоевая или тиоктовая кислота локализуется во всех средах организма: клеточных мембранах, цитоплазме и внеклеточной жидкости.
Наиболее активными из группы ферментов являются металлопротеины, обладающие активностью супероксидцисмутазы. В зависимости от содержания металлов супероксидцисмутазы (СОД) подразделяются на СОД, содержащие медь и цинк (Сu и Zn-Cu/Zr- СОД); СОД, содержащие марганец (Мn-Mn-СОД); СОД, содержащие железо (Fe-Fe-СОД).
Рис. 7.
Участие витаминов Е и С, а-липоевой кислоты в инактивации свободных радикалов.
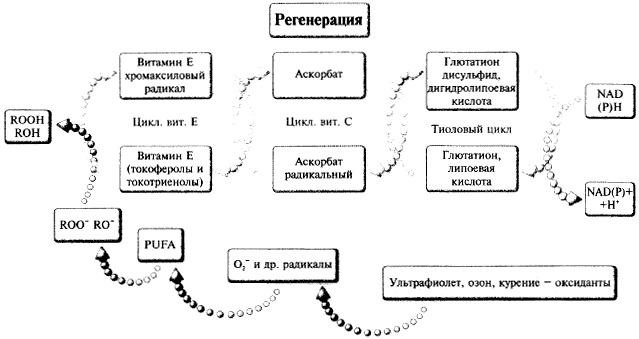
Липоевая кислота имеет дитиолановое кольцо (окисленная форма), которое разрушается при восстановлении, переходит в дигидролипоевую кислоту (восстановленная форма) или липоат.
СОД, содержащая медь и цинк, локализуется в растворимой фракции клеток, а содержащая марганец — в митохондриях. Функция суперксиддистутазы заключается в восстановлении О2*- доН2О2. Указанная реакция протекает
2O2*- + 2Н+ -супероксиддисмутаза-> Н2О2 + О2
Образовавшаяся в процессе реакции перекись водорода затем разлагается при участии фермента каталазы до воды и кислорода:
2Н2О2 –каталаза-> 2Н2О + О2
Указанная реакция является своего рода «первой линией» защиты от избытка свободных радикалов и, как правило, обеспечивает обезвреживание свободных радикалов в клетке. В том же случае, когда эта защитная мера бывает недостаточной и происходит образование НО', который осуществляет окисление полиненасыщенных жирных кислот (PUFA), то такие липидные радикалы вступают во взаимодействие с естественным антиокдидантом — витамином Е. Адекватная функция антиоксидантной системы поддерживается согласованной активностью цикла витамина Е, цикла витамина С и цикла а-липоевой (тиоктовой) кислоты или тиоловым циклом (рис. 7).
К настоящему времени почти полностью установлен механизм участия свободных радикалов и патогенезе сосудистых осложнений. Схематически это представлено на рис. 8. Свободнорадикальные соединения (О2- и ONOO-) активируют скорость апоптоза, окисление ЛПНП и образование ядерного транскрипционного фактора NF-kb. Последний с участием нескольких белков (сс-фактор некроза опухолей, интерлейкин, II- 1р, молекулы адгезии) опосредует механизмы, способствующие тромбогенной трансформации сосудистой стенки (рис. 9).
Рис. 8.
Образование и участие свободных радикалов в патеногенезе сосудистых осложнений.
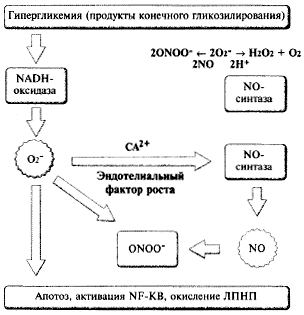
Рис. 9.
NF-kB опосредованные механизмы, ведущие к тромбогенной трансформации сосудистой стенки.
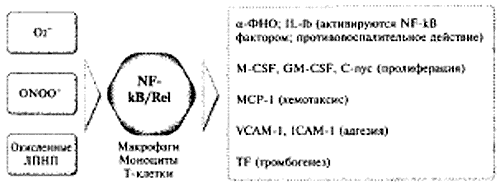
Факторы роста: M-CSF-моноцит колонистимулирующий;
GM-CSF - гранулоцит-моноцит колонистимулирующий;
МСР-1 моноцит хемоаттрактный белок;
VCAM-1 - белок 1 адгезии сосудистых клеток;
ICAM-1 - внутриклеточная адгезивная молекула-1;
TF - тканевой фактор.
Дефицит инсулина или инсулиновая резистент-ность, являющиеся причиной сахарного диабета, способствуют развитию окислительного стресса, который в свою очередь прямо и опосредованно приводит к развитию сосудистых осложнений рис. 10.
В организме активность ПОЛ и активность системы антиоксидантной защиты находятся в постоянном равновесии, которое у больных СД нарушено, и скорость перекисного окисления липидов превышает способность антиоксидантной системы гасить избыточное количество свободных радикалов. Это обстоятельство диктует необходимость применения антиоксидантов в комплексной терапии СД. Наши исследования и данные других авторов показывают, что применение различных антиоксидантов (витамина Е, С, никотинамида, диквертина, тиоктацида и др.) снижает содержание продуктов ПОЛ (диеновые конъюгаты и малоновый диальдегид), повышает активность ферментов антиоксидантной защиты, что оказывает положительное воздействие на стабилизацию сосудистых осложнений диабета, сохраняя работоспособность и качество жизни больных диабетом, что в конечном итоге отдаляет время инвалидизации и снижает летальность.
Рис. 10.
Осложнения сахарного диабета.
|
Дефицит инсулина или инсулинрезистентность | ||||
|
↓ |
↓ |
↓ |
↓ |
↓ |
|
Истощение антиоксидантов |
Гликирование белков |
↑ Свободных радикалов |
Синтез простаноидов |
Полиоловый путь |
|
↓ |
↓ |
↓ |
↓ |
↓ |
|
ОКИСЛИТЕЛЬНЫЙ СТРЕСС | ||||
|
↓ | ||||
|
АКТИВАЦИЯ NF-kB | ||||
|
↓ |
↓ |
|
↓ |
↓ |
|
НЕФРОПАТИЯ антиоксидантов |
НЕЙРОПАТИЯ белков |
|
РЕТИНОПАТИЯ |
АНГИНОПАТИЯ |



