Flupirtine in Pain Management
Статьи Опубликовано в журнале:CNS Drugs »» 2010; 24 (10) Adis Data Information BV.
Pharmacological Properties and Clinical Use
Jacques Devulder
Department of Anaesthesia, Section Pain Clinic, Ghent University Hospital, Ghent, Belgium
| Abstract |
Flupirtine is a centrally acting, non-opioid analgesic that is available in a number of European countries for the treatment of a variety of pain states. The therapeutic benefits seen with flupirtine relate to its unique pharmacological properties. Flupirtine displays indirect NDMA receptor antagonism via activation of potassium channels and is the first representative of a pharmacological class denoted the ‘selective neuronal potassium channel openers’. The generation of the M-current is facilitated by flupirtine via the opening of neuronal Kv7 potassium channels. The opening of these channels inhibits exaggerated neuronal action potential generation and controls neuronal excitability. Neuronal hyperexcitability is a physiological component of many pain states such as chronic pain, migraine and neurogenic pain. |
Flupirtine is a centrally acting, non-opioid analgesic that belongs to the triaminopyridine class. Its chemical structure, mode of action and overall drug characteristics differ distinctly from those of other analgesics on themarket. Flupirtine was synthesized by Degussa Pharma (Frankfurt/Main, Germany) and was first approved in the 1980s in Germany.
Flupirtine is approved in a number of European countries (Germany, Italy, Portugal, Russia, Slovakia, Estonia, Latvia, Lithuania), and also Brazil, to treat various pain states.[1] Although its approved indications differ between countries, they include the clinical management of musculoskeletal pain, neuralgias and neuritis, headache, dysmenorrhoea and postoperative, cancer and trauma pain.
In addition to its use in pain management, flupirtine displays muscle relaxant and anticonvulsant activity.[1] The unique pharmacological properties of flupirtine contribute to its therapeutic benefits, without worrisome adverse effects such as respiratory depression, tolerance and dependence that are typical of opioids, or the gastrointestinal and renal problems associated with nonsteroidal anti-inflammatory drugs (NSAIDs).[1] Flupirtine displays properties that are different to common analgesics and is the first representative of an entirely different class of analgesics denoted by Kornhuber et al.[2] as ‘selective neuronal potassium channel openers’ (SNEPCO).
The purpose of this review is to describe the pharmacological properties of flupirtine, compare them with other analgesics and examine how these properties contribute to its efficacy in the management of pain. Studies used in this review were identified via the MEDLINE database using the terms: ‘flupirtine’ OR ‘flupirtine maleate’ AND ‘pain’. All years of publication, up to March 2009, were included, as were non-English references. Reference lists of identified publications were also scanned for articles relevant to the topic. Only clinical studies of standard flupirtine formulations (i.e. those containing flupirtine alone) were included.
1. Mechanism of Action and Pharmacological Properties
The antinociceptive activity of flupirtine is similar to that of opioid agonists and mixed agonistantagonists but its mechanism of action is not based on the opioid mechanism.[3] The morphine antagonist, naloxone, did not inhibit the analgesic activity of flupirtine. Furthermore, flupirtine did not demonstrate any binding affinity for µ, δ or k opioid receptors in the rat brain.[3] Initially, the mode of action of flupirtine was not well understood and early results suggested that part of the antinociceptive activity of the drug may be exerted via the descending noradrenergic painmodulation system,[4] as α-adrenoceptor inhibitors significantly reduced the antinociceptive activity of flupirtine (p<0.05). However, direct binding studies revealed that flupirtine did not display any affinity for α1- or α2-adrenoceptors, or serotonin 5-HT1 or 5-HT2 receptors.[4]
Further analyses revealed that flupirtine only displays weak modification of prostaglandin formation and therefore lacks clinically significant anti-inflammatory activity.[5] Flupirtine significantly inhibited basal prostaglandin I2 in rat aortaand arachidonic acid-stimulated prostaglandin release in perfused guinea pig heart (concentration of drug producing 50% inhibition 5–50 µmol/L).
A dose-dependent inhibition of collagen-induced human platelet aggregation was also observed with flupirtine. Additionally, flupirtine inhibited adenosine diphosphate-induced aggregation of human platelets but this was not dose dependent. Flupirtine was one order of magnitude less potent than indomethacin throughout these tests. Moreover, flupirtine (<23 µmol/L) failed to inhibit arachidonic acid-induced thromboxane formation and release from human platelets.[5]
Flupirtine also displays indirect NDMA receptor antagonism via the activation of potassium (K+) channels.[6] Initially, it was thought that flupirtine properties were consistent with that of NDMA receptor antagonism. However, no interaction with the binding sites of theNDMAreceptor could be found when direct studies were performed with flupirtine.[7] Furthermore, flupirtine did not display any influence on NMDA-induced ion currents during patch-clamp investigations,[6,7] and no affinity for NMDA receptor binding sites was seen in human post-mortem brain tissue.[6]
Jakob and Krieglstein[7] demonstrated that the administration of flupirtine activated G-proteinregulated inwardly rectifying K+ (GIRK) channels in rat hippocampal neurons. GIRK channels characterize a new family of K+ channels that are distinctive from the classic voltage-dependent K+ channels.[6] GIRK channels contribute to the maintenance of resting membrane potential and manage the excitability of the cell. The conductance of these channels is characterized by an increased current during hyperpolarization and a decreased current during cell depolarization.[7] An outward conductance of K+ ions stabilizes the resting membrane potential close to K+ equilibrium potential.[6] The activation of GIRK channels results in the hyperpolarization of the neuronal membrane.[7] Therefore, flupirtine stabilizes the membrane potential indirectly by activating GIRK channels.
Despite being a well established drug, it has been suggested that flupirtine is the first among a new class of substances called SNEPCO.[2] Flupirtine, along with retigabine (an analogue of flupiritine), is a neuronal Kv7 activator.[8,9] The Kv7 potassium channel family has five members (Kv7.1–Kv7.5), with Kv7.2–Kv7.5 specifically referred to as neuronal.[ 8] These channels facilitate the generation of the M-current and control neuronal excitability.[10] The M-current was identified as a membrane hyperpolarizing current activated by muscarine; a decreased conductance to K+ results from cholinergic muscarinic receptor activation. The M-current is a low-threshold, non-inactivating, voltage-dependent K+ current that limits repetitive firing of neuronal action potentials.[2,9] Essentially, the presence of flupirtine attenuates the generation of neuronal action potentials.[2]
The pathophysiology of several disease states such as epilepsy, migraine and chronic pain includes a component of neuronal hyperexcitability.
Under these conditions, hyperpolarization of the resting membrane potential and hence decreased action potential generation by K+ currents, as seen with flupirtine, may provide a unique therapeutic approach in themanagement of diseases associated with neuronal hyperexcitability.[9,10]
2. Pharmacokinetic Properties
2.1 Absorption
Flupirtine is a water-soluble compound that is rapidly absorbed from the gastrointestinal tract.[1,11] Following oral administration of flupirtine 100mg, the maximum (peak) plasma drug concentration (Cmax) was 0.77mg/L in healthy volunteers, 1.12 mg/L in elderly patients and 0.72 mg/L in patients with renal impairment (table I).[12] Flupirtine 200 mg resulted in a Cmax of 1.98 mg/L in healthy young volunteers. Rectal administration of flupirtine 150 mg produced a Cmax of 0.89 mg/L after 5.7 hours.[13]
Absolute bioavailability for flupirtine 80mg was 90.0% and 72.5% for oral and rectal administration, respectively, when compared with intravenous administration.[13] Significant linear dose-dependent plasma concentration increments were observed with flupirtine in healthy young volunteers.[14]
2.2 Distribution
In healthy volunteers, the apparent volume of distribution for oral flupirtine 100 mg is 154L. Renally impaired and elderly patients recorded volumes of distribution of 212 L and 195 L, respectively.[12] Binding of flupirtine to human plasma proteins is concentration independent and about 80%, and hence, classified as low.
2.3 Metabolism and Elimination
Flupirtine is bio-transformed in the liver into two primary metabolites, 4-fluoro-hippuric acid and the N-acetylated analogue D13223, which is pharmacologically active (about 30% of the analgesic potency of the parent drug).[8,15] Themetabolic steps to formation of D13223 include hydrolysis of the urethane structure and subsequent N-acetylation.
Table I. Pharmacokinetic properties of oral flupirtine 100mg[12]
| Parameter | Healthy volunteers |
Elderly patients |
Renal impairment |
| AUC (mg*h/L) | 6.07 | 10.34 | 6.66 |
| CL (mL/min) | 275 | 161 | 263 |
| Cmax (mg/L) | 0.77 | 1.12 | 0.72 |
| t½ (h) | 6.50 | 14.00 | 9.80 |
| tmax (h) | 1.60 | 1.80 | 1.80 |
| Vd (L) | 154 | 195 | 212 |
| AUC= area under the plasma concentration-time curve; CL = total body clearance; Cmax = maximum (peak) plasma drug concentration; t½ = elimination half-life; tmax = time to reach Cmax; Vd = volume of distribution |
|||
p-Fluoro-hippuric acid was identified as a further metabolite. A prerequisite for the formation of this metabolite is the hydrolysis of the original p-fluorobenzyl structure to form p-fluoro-phenyl-methanol. Subsequent oxidative cleavage results in the intermediate p-fluoro-benzoic acid, which is then subjected to glycin-conjugation to form p-fluoro-hippuric acid. This moiety is pharmacologically inactive.[1,8,15]
Following administration of oral flupirtine 100mg, the mean terminal plasma elimination halflife is about 6.5 hours in healthy subjects, 9.8 hours in patients with mild renal impairment and 14.0 hours in elderly patients;[12] corresponding values in healthy adult subjects upon intravenous and rectal administration are 8.5 and 10.7 hours, respectively.[13] The clearance of flupirtine is 275mL/min in healthy subjects, 263mL/min in patients with renal impairment and is decreased to 161mL/min in elderly patients (table I).[12] Flupirtine is predominantly excreted via the urine (72%).[13]
Methling et al.[8] performed in vitro metabolism analyses of flupirtine and determined that flupirtine does not undergo cytochrome P450 enzymemediated metabolism to a significant extent. This group also concluded that human monoamine oxidase isoenzymes were unlikely to participate in in vivo metabolism of flupirtine. In contrast, human myeloperoxidase and horse radish peroxidase enzymes produced rapid turnover of flupirtine, suggesting that flupirtine was an excellent substrate for these enzyme pathways.[8]
Currently, there are no published data highlighting interactions between flupirtine and other drugs.
3. Formulations, Dose and Administration
These are summarized in table II.[1,16,17]
4. Pain Indications
Flupirtine can be useful in the treatment of awide range of pain states. In line with its mechanism of action promoting neuronal rest, it has been proven useful in conditions involving neuronal hyperexcitability such as chronic pain (non-malignant and malignant), migraine and neurogenic pain.[3,18-22]
Furthermore, its effect as a muscle relaxant represents an added value in pain conditions associated with increased muscle tension such as musculoskeletal back pain,myofascial pain and tension headache.[18,22-24] Flupirtine is also beneficial in the short-term treatment of acute to moderate painsuch as postoperative pain, trauma and dysmenorrhoea.[25] The approved indications of flupirtine differ between countries but mainly include the clinical management of musculoskeletal pain, postoperative pain, headache, dysmenorrhoea, neuralgia and neuritis, post-traumatic pain (trauma and chemical burns) and cancer pain.[16,17]
Table II. Route of administration, dose preparations and recommended dosage of flupirtine[16,17]
| Route of administration |
Dose preparations (mg) | Recommended dosage | Maximum daily doses |
|
| adults | children (aged >6 y) | |||
| Oral | 50 and 100 | 100 mg three to four times daily a | 50 mg three to four times daily | 6 |
| Rectal | 75 and 150 | 150 mg three to four times daily | 75 mg three to four times daily | 6 |
| a For more severe pain, an oral 200 mg dose may be taken up to three times daily. | ||||
5. Analgesic Properties
5.1 Preclinical Studies
Following electrostimulated pain and hot plate tests in mice and tooth pulp stimulation tests in dogs, it was determined that flupirtine displays central antinociceptive activity, as the effects seen with flupirtine administration were distinctive, with centrally acting analgesic properties.[3] Flupirtine was more potent than paracetamol, as potent as pentazocine and less potent than morphine and buprenorphine in the electrostimulated pain test performed in mice. In the hot plate test, flupirtine (dose that produces a 50% effective response [ED50] 32mg/kg orally) was half as potent as morphine (ED50 1.5 mg/kg orally).[3] The electrical tooth pulp stimulation test in dogs revealed that the ED50 of flupirtine was 3.5mg/kg and 0.7 mg/kg for oral and intravenous administration, respectively.[3]
Flupirtine produced a significant increase in morphine antinociception when the two drugs were administered in combination in two rat models of pain: carrageenan paw inflammation and streptozocin-induced diabetic neuropathy.[26]
Flupirtine 5 or 10mg/kg in combination withmorphine 0.4 mg/kg produced a significant reversal of carrageenan-induced hyperalgesia (p<0.001) that was similar to the effect seen with morphine 1.6 mg/kg (p<0.05), i.e. the antinociceptive activity of morphine was increased 4-fold by flupirtine.
When administered as monotherapies, flupirtine 10mg/kg and morphine 0.4mg/kg were ineffective in reducing the hyperalgesia caused by carrageenan; however, when administered as combination therapy they resulted in complete antinociception (p<0.05). In streptozotocin-induced diabetic neuropathic rats, flupirtine 10mg/kg, alone or in combination with morphine 1.6 kg/mg, produced significant antinociception (p<0.001) and complete reversal of streptozotocin-induced hyperalgesia (p<0.05); significantly greater antinociception was seen with the combination when compared with flupirtine 10 mg/kg alone.[26]
Flupirtine has also demonstrated potent muscle-relaxant properties in animal studies. For example, a study in conscious rats compared the effect of several analgesics with flupirtine on muscle tone. At doses comparable to antinociceptive doses, flupirtine reduced muscle tone without inducing sedative effects such as ataxia and without reducing spontaneous motor activity.[27]
5.2 Clinical Studies
Flupirtine has demonstrated efficacy in a range of pain conditions including musculoskeletal pain (e.g. lower back pain, pain due to osteoporosis), headache, neurogenic pain and cancer pain. A summary of placebo- and active-controlled clinical studies is presented in table III. The majority of studies included in this section enrolled only a relatively small number of patients. Data from small controlled studies and open-label studies are discussed in this section because largescale studies are lacking. For certain studies, results are to be interpreted with caution because of the inherent limitations of their open-label design and/ or small sample size.
The potential analgesic efficacy of flupirtine was suggested in a pharmacodynamic study in healthy adult subjects, where pain induced via carbon dioxide stimulation of the nasal mucosa was reduced by flupirtine in a dose-dependent manner.[14] Flupirtine 200mg produced greater changes in carbon dioxide-induced somatosensory evoked potentials than a 300 mg dose. An interesting point to note from this study is that an increase of approximately 10mmHg (p<0.05) in systolic blood pressure was seen following flupirtine 100 and 200 mg administration.[14]
Table III. Design and results of placebo- and active-controlled flupirtine (FLU) studies
| Study | Design | No. of patients | Drugs and dosage | Duration | Pain type | Outcome measures | Efficacy |
| Placebo-controlled | |||||||
| Worz et al.[24] | db, mc | FLU (n = 30); PL (n = 23) |
FLU 100mg tid | 2wk run-in phase; 2wk treatment phase |
Chronic tension headache | HPAL; global assessments of efficacy and tolerability |
Significant reduction in pain intensity (p = 0.013), headache occurrence (p = 0.019) and pain severity (p = 0.030) vs PL |
| Active-controlled | |||||||
| Li et al.[20] | r, db | FLU (n = 105); TRA (n = 104) |
FLU 100mg tid TRA 50mg tid |
1wk | Sub-acute lower back pain |
11-point numerical rating scale; pain relief rate |
Significant reductions in pain intensity (p < 0.0001) for both drugs |
| Worz et al.[22] | r, db, mc | FLU (n = 54); CHL (n = 57); PL (n = 55) |
FLU 100mg qid CHL 200mg qid |
1wk wash-out phase; 1wk treatment phase |
Musculoskeletal lower back pain |
Global scale assessments |
Differences in responder rates for pain intensity insignificant for FLU vs CHL and PL; significant difference in global assessment of efficacy for FLU vs PL (p < 0.007) but not for FLU vs CHL (p = NS) |
| Luben et al.[19] | db | FLU (n = 35); TRA (n = 36) |
FLU 100mg qid TRA 50mg qidicant |
4wk | Cancer pain | SPID; 5-point verbal scale; global assessments |
Pain reduction not significantly better with FLU than TRA |
| Sitzer[28] | sb | FLU (n = 28); ASP (n = 30) |
FLU 100mg tid ASP 500 mg tid |
5 d treatment phase | Spinal root irritation syndrome |
4-point pain severity scales |
Lower pain scores and less restriction in walking for FLU vs ASP (p =NS) fewer GI symptoms with FLU vs ASP (p = NS) |
| Mastronardi et al.[29] | db | FLU (n = 20); DICL (n = 20) |
FLU 100mg qid DICL 50mg qid |
1 d | Orthopaedic postoperative pain |
VAS | Significant pain alleviation for FLU vs DICL after third dose (p = 0.02) |
| Marczyk[30] | r, op | FLU (n = 31); DICL (n = 30) |
FLU 100mg tid DICL 50mg tid |
1wk | Musculoskeletal pain | 10-point VAS; global assessments |
Similar reduction in pain FLU vs DICL |
| Million et al.[31] | r, db | FLU (n = 20); PARA (n = 20) |
FLU 100mg qid max PARA 1000mg qid max |
5 d treatment phase | Migraine | 4-point verbal rating scale |
Similar analgesic consumption FLU vs PARA; lower number of AEs reported for FLU vs PARA (p = NS) |
| ASP= aspirin; AE= adverse event; CHL= chlormezanone; db = double-blind; DICL = diclofenac; GI = gastrointestinal; HPAL= Hamburg Pain Adjective List; max= maximum; mc = multicentre; NS = not significant; op = open-label; PARA = paracetamol; PL = placebo; qid = four times daily; r = randomized; sb = single-blind; SPID = sum of pain intensity differences; tid = three times daily; TRA = tramadol; VAS = visual analogue scale. | |||||||
5.2.1 Musculoskeletal Pain
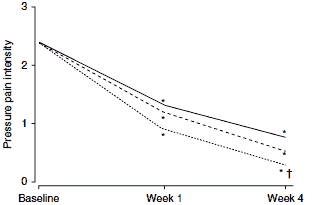
In a postmarketing surveillance study involving 7806 patients treated under general practice conditions, flupirtine was an effective analgesic for the control of acute, sub-acute and chronic musculoskeletal pain.[18] This study provides evidence of the efficacy of flupirtine in a realworld setting, although the open-label nature of the study should be considered when assessing study results. Nevertheless, clinically and statistically significant beneficial effects on pain intensity (figure 1a), pressure pain intensity (figure 1b) and muscle tenseness (figure 1c) were observed within 1 week of the initiation of flupirtine therapy (200–300 mg/day in the majority of patients).
After 1 week of therapy the response rate (patients with a >1-point improvement on the 5-point visual analogue scale) was 94%, 89.4% and 85.9% for patients with acute, sub-acute and chronic pain, respectively. After 4 weeks of therapy, the response rate was >95% in all patient groups. The effectiveness of flupirtine for controlling pain was reflected in statistically significant and clinically relevant improvements in restrictions in daily life and sleep disorders, both indicators of quality of life. Flupirtine was well tolerated in this study. The overall incidence of adverse effects was 0.9%, and no serious events were reported. Tiredness and dizziness were the most common adverse events.[18]
 |
| a |
 |
| b |
 |
| c |
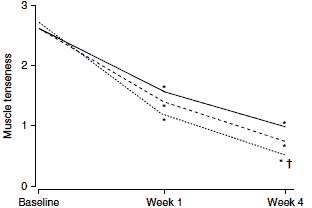 |
| Fig. 1. Effects of flupirtine in a postmarketing surveillance study on (a) pain intensity; (b) pressure pain intensity; and (c) muscle tenseness. These were measured on a 5-point visual analogue scale from 0= no pain/tenseness to 4= very strong pain/tenseness (reproduced from Mueller-Schwefe,[18] with permission). * p<0.01 for change compared with baseline; † p<0.01 for difference between acute and chronic pain. |
A randomized, double-blind study revealed that flupirtine was comparable to tramadol in terms of pain reduction in patients (n = 209) with sub-acute lower back pain.[20] Both flupirtine and tramadol significantly (p<0.0001) reduced pain intensity following a 1-week treatment cycle.
Good or excellent ratings for the physician’s global efficacy assessments (flupirtine 63.8% vs tramadol 64.4%; p= 0.633) and functional capacity (64.8% vs 64.4%; p= 0.481) were similar in the two groups.[20] A placebo control group was not included and would have allowed for assessment of a placebo response, which commonly occurs in this patient population.
Flupirtine is a potent muscle relaxant and is therefore highly effective in painful muscle tension. For example, in a randomized, double-blind study in patients (n = 166) with chronic musculoskeletal low back pain, the muscle relaxant effect of flupirtine <400 mg/day (60.9% response rate) was comparable to that of chlormezanone <800 mg/day (47.8%) and placebo (43.8%).[22] Although not achieving statistical significance in terms of superiority, the response rate for pain intensity was numerically higher in flupirtine recipients (54.3%) compared with chlormezanone (45.7%) and placebo recipients (33.4%). Corresponding response rates for tension were 47.8%, 43.4% and 33.4%. For the global assessment of efficacy, the difference between flupirtine and placebo was statistically significant (p<0.007).[22]
Furthermore, in a prospective standardized evaluation of patients with chronic treatment-refractory back pain, 2 weeks of treatment with flupirtine significantly improved all muscle-specific indicators, including pain pressure threshold (+48%), pain pressure tolerance (+27%) and depth of penetration in the muscle (+18%) [all p<0.001]. A clinically observable and statistically significant relief of pain was also observed.[32]
Flupirtine displayed similar clinical analgesic efficacy to diclofenac in a small double-blind study involving orthopaedic patients.[29] Patients received either flupirtine 100 mg (n = 20) or diclofenac 50 mg (n = 20) up to a maximum of four doses per day following surgery; pain intensity was evaluated at 30 minutes and 60 minutes following the first dose and every 60 minutes following the subsequent doses using a visual analogue scale. No significant changes in pain reduction were seen with either drug during the first 2 hours following administration of the first dose. Diclofenac produced significant pain reduction 30 minutes after the second dose (p = 0.01), while a significant reduction in pain was seen 2 hours after the second dose with flupirtine (p = 0.001). There were no significant differences observed between the two drugs in terms of pain alleviation at any of the time intervals after the second dose. However, after the third dose, pain reduction significantly favoured flupirtine (p = 0.02).[29]
Flupirtine also showed similar efficacy to diclofenac in a comparative study in 61 patients with pain of musculoskeletal origin.[30] Following treatment with flupirtine 100 mg three times daily (n= 31) or diclofenac 50mg three times daily (n= 30) for a minimum of 7 days, physician’s assessment of pain at rest and during body movement, local sensitivity, level of mobility and sleep disturbance showed improvements in all parameters in both treatment groups with no significant betweengroup differences. Similarly, according to patient self-assessments using a 10-point visual analogue scale, both flupirtine and diclofenac treatment resulted in statistically significant improvements in all parameters at study end compared with baseline (p<0.001). Also, at the end of the study, physician-rated improvements in global assessment of efficacy was excellent or good for 77.5% of flupirtine recipients and 86.7% of diclofenac recipients; the corresponding figures for patient ratings were 80.6% and 90.0%, respectively.[30]
Flupirtine (mean dosage 270mg/day) provided effective analgesia in an open-label, prospective, observational study conducted in elderly patients with pain secondary to osteoporosis (n = 869), with a reduction in pain of 43% for the lower back, 44% for the back, 40% for the arm and shoulder and 40% for all other pain states (all p 5.2.2 Headache
In patients who could not tolerate or had insufficient response to conventional analgesics (n = 50), flupirtine effectively relieved pain for 2–5 hours in those with chronic tension headache or cervicogenic headache in a small, open-label, prospective study.[23] A good level of analgesic activity was also observed in patients with CNS lesions with secondary insertion tendinopathies, myalgia and (in some cases) trigger point syndromes. The effect in patients with fibromyalgia (which does not respond well to treatment) was less marked, but pain was relieved by 20–70% in six patients.[23] In a comparative double-blind study, flupirtine treatment was associated with lower pain scores, less confinement to bed and restriction in working ability than patients treated with paracetamol for acute migraine attacks.[31] Patients received oral flupirtine 100 mg (n = 20) or oral paracetamol 1 g (n = 20) with a maximum of four doses per day for 5 days. Total analgesic consumption was similar among the two groups (6.65 vs 6.85 doses, respectively). Adverse effects were minor and infrequent in both groups; however, a lower number of adverse effects were reported among flupirtine patients when compared with patients receiving paracetamol.[31]
Flupirtine treatment produced superior reductions of pain symptoms in patients with chronic tension headache in a small placebo-controlled study.[24] Following a run-in period, patients were administered flupirtine 100 mg (n = 30) or placebo (n = 23) three times daily for a duration of 2 weeks.
A significant reduction in pain intensity was observed with flupirtine versus placebo (p = 0.013). In addition, headache occurrence and average pain severity were significantly improved in flupirtinetreated patients compared with placebo (p= 0.019 and p = 0.030, respectively). Non-significant improvements in sleep disturbance, tenderness and tension were greater with flupirtine than placebo.
A global assessment of tolerability by the treating doctor demonstrated that tolerability was ‘‘very good’’ or ‘‘good’’ in 84% of flupirtine-treated patients and 72%of patients receiving placebo.[24]
5.2.3 Neurogenic Pain
Greater short-term pain relief was observed with flupirtine (mean dosage 380 mg/day) when compared with aspirin (mean dosage 1800mg/day) in patients with lumbar and cervical spinal root irritation (n = 58) in a small single-blind study.[28]
A patient assessment of pain intensity at study end revealed that 25% of flupirtine-treated patients reported no pain, 46% reported an improvement, 29% had no improvement at all and no patients reporting a deterioration compared with 35% of patients treated with aspirin who reported an improvement, 62% remained unchanged and deterioration reported by one patient. A total of 21% of flupirtine-treated patients reported gastrointestinal symptoms compared with 34% of patients treated with aspirin (difference not statistically significant).[28]
5.2.4 Cancer Pain
A small comparative multicentre study (n = 71) determined that flupirtine reduced pain more effectively than tramadol in cancer patients with pain symptoms; however, this difference did not reach statistical significance.[19] A global assessment by the treating doctor of the success of the analgesic therapy showed that ‘‘good’’ and ‘‘very good’’ were applied to 62% of flupirtine-treated patients and 46% of tramadol-treated patients (p = 0.230, Fisher’s two-sided test).[19] Flupirtine was also associated with more ‘‘good’’ or ‘‘very good’’ results when compared with pentazocine (68% vs 50% on a 4-point verbal grading scale) in the treatment of patients with cancer pain in another small study; however, the difference observed also did not reach statistical significance (p>0.3).[33] In this trial, patients with ‘‘severe’’ to ‘‘very severe’’ cancer pain (n = 52) received oral flupirtine 100 mg or pentazocine 50 mg with a maximum daily dose of six capsules for 1 week. Baseline pain and dosage were similar between the two groups. Both groups had a similar incidence of adverse reactions; however, an increased incidence of CNS adverse events was seen in pentazocine-treated patients compared with patients receiving flupirtine.[33]
5.3 Combination Therapy
In a pilot open-label, dose-escalation study, flupirtine reduced neuropathic pain associated with cancer in patients concurrently taking opioid medication (n = 10).[34] Initially patients were administered flupirtine 100 mg four times daily and the dose could be increased by 100 mg four times daily every 2 days to a maximum of 300 mg four times daily; dose increases were at the physician’s discretion. Significant reductions in neuropathic pain discriminant scores, average pain and percentage pain relief (p<0.01) were seen with flupirtine without an increase in adverse effects.[34]
6. Anti-Apoptotic, Antioxidative and Cytoprotective Properties
Chronic pain leads to nerve degeneration,[35] and an analgesic with neuroprotective properties in addition to pain relief would be an advantage in the treatment of chronic pain.
Flupirtine displays potent antioxidant properties in rat brain mitochondria and phaeochromocytoma 12 cell culture.[36] The administration of flupirtine significantly inhibited free radical reactions; this effect was concentration dependent. It was more potent than nimodipine, a dihydropyridine calcium antagonist, and as effective as dopamine or desferoxamine. In addition, protein carbonyl formation was reduced and survival of hydrogen peroxide-induced injury was improved with flupirtine administration.[36]
Flupirtine effectively prevented death by apoptosis and reduced formation of reactive oxygen species (ROS) in cultured human retinal pigment epithelial (RPE) cells.[37] When RPE cells were subjected to 72 hours of experimental ischaemia, flupirtine prevented apoptosis by a mode of action that does not involve NMDA receptors.[38]
Glutathione depletion is associated with a failure to maintain ROS levels and, inevitably, with cell death by apoptosis.[38] Flupirtine maintains glutathione levels and hence prevented cell death in human RPE cells in the study performed by Wood et al.[38]
Marked neuroprotection was seen with flupirtine in rat organotypic hippocampal slice cultures when neurodegeneration was induced via NDMA exposure, oxygen and glucose deprivation or serumwithdrawal.[39] It was also determined that flupirtine provides neuroprotection as addon therapy in a rat model of autoimmune optic neuritis.[40] A significant increase in survival in retinal ganglion cells (RGCs), neurons that form the optic nerve, was observed with flupirtine treatment. An improvement in visual function in the acute phase of optic neuritis was seen and RGCs were protected from degeneration under noninflammatory conditions with flupirtine therapy.
The effects produced by flupirtine therapy did not involve the anti-apoptotic Bcl-2 mechanism.[40]
In vivo cytoprotection at a mitochondrial level in rat hearts was demonstrated with flupirtine.[41] Flupirtine was associated with a significant increase in mitochondrial calcium uptake compared with the control (p<0.01), which suggests that it protects against ischaemic damage.[41]
7. Flupirtine Use in Other Disease States
Flupirtine has shown some success in the treatment of fibromyalgia symptoms in patients diagnosed with the disease.[42] Currently, there are three drugs that are approved by the US FDA for the management of fibromyalgia, a disease that is characterized by musculoskeletal pain, fatigue and depression. These include pregabalin,[43] duloxetine[44] and milnacipran.[45] Flupirtine therapy was associated with an improvement in all symptoms associated with the disease not only pain symptoms in an open-label case series report.[42] The report describes the use of flupirtine in the treatment of fibromyalgia pain in four patients in whom other treatment strategies had previously failed. Full relief from pain was achieved in three of the four patients and they remained fibromyalgia symptom free from 5 to >18 months.
One subject achieved mild relief from pain.[42] In 2008, the US FDA approved a randomized, double-blind, placebo-controlled, phase II clinical trial investigating oral preparations of flupirtine for the treatment of fibromyalgia; no results have been released to date.
8. Tolerability
The most common adverse events occurring during flupirtine therapy include drowsiness, dizziness, heartburn, headache, dry mouth, fatigue and nausea[25,46-48] (table IV). Such events tend to be mild and transient,[48] and are most likely to occur in the first 6 months of therapy.[47] No clinically significant alterations in laboratory parameters or vital signs, including blood pressure, heart rate, ECG, renal function, haematologic and metabolic parameters, have been observed during flupirtine therapy.[25,46-48] However, a small number of patients have experienced abnormalities in liver function tests (see below).
Table IV. Most common adverse events reported with flupirtine use
| Symptoms | Frequency (%) |
| Dizziness | 9–19[46,47] |
| Nausea | 4–14[25,46,48] |
| Sleep disturbances | 10[46] |
| Headache | 4–5.4[46] |
| Drowsiness | 5–25[46-48] |
| Fatigue | 8[25] |
Compared with other available analgesic options, flupirtine is a well tolerated choice of therapy. For example, the use of NSAIDs is limited by gastrointestinal adverse events, including dyspepsia, which has been reported to occur in up to 40% of NSAID recipients.[49] Notably, in a randomized, open-label study of 61 patients withmusculoskeletal disorders, tolerability was considered excellent or good by 2-fold more flupirtine than diclofenac recipients (100% vs 50%; p< 0.01) and statistically significantly less flupirtine than diclofenac recipients experienced adverse events (16% vs 63%; p< 0.001), with more cases of gastrointestinal adverse events occurring among diclofenac recipients (figure 2).[30]
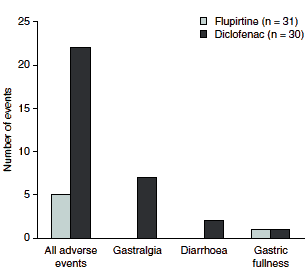 |
| Fig. 2. Adverse events in a comparative study of flupirtine and diclofenac for the treatment of musculoskeletal diseases (n = 61).[30] |
NSAID-induced upper gastrointestinal symptoms have an important impact on health-related quality of life. It has been reported that more than 50% of NSAID recipients were at least ‘‘fairly bothered’’ by gastrointestinal symptoms during therapy.[50] NSAID use has also been associated with a risk of endoscopically diagnosed ulcers, symptomatic ulcers and serious upper gastrointestinal events, including bleeding, perforation and obstruction.[51] Thus, while NSAIDs may offer efficacy advantages over flupirtine in the treatment of inflammatory pain syndromes or migraine, flupirtine has clear tolerability advantages over NSAIDs and therefore represents an excellent alternative in patients at risk of NSAIDassociated gastropathy and in pain states that do not have a particular inflammatory pathophysiological component.
The tolerability profile of flupirtine is also superior to that of opioids, which can cause constipation, nausea, vomiting, sedation, confusion, pruritus, urinary retention and respiratory depression, and are associated with the development of tolerance and dependence.[52] In contrast, no signs of dependence or tolerance were observed with flupirtine treatment for chronic pain in a 12-month study.[46] Oral flupirtine 100mg three times daily was significantly better tolerated than tramadol 50 mg administered three times daily in a double-blind study.[20] Significantly fewer of the adverse effects that are usually associated with opioids, such as nausea, vomiting, dizziness and sedation, were observed with flupirtine administration (33%vs 49%; p = 0.02).[20] In addition, significantly fewer flupirtine-treated patients dropped out due to treatment-emergent adverse events than tramadol recipients (1% vs 15%; p< 0.001).[20]
In another direct comparison with tramadol, the rate of adverse events was markedly lower in flupirtine recipients (5.7% vs 19.4%), and there were no unwanted opioid-induced effects in the flupirtine group (figure 3).[19] Flupirtine was also better tolerated than the opioid pentazocine in an analysis of data from two multicentre trials. Nausea, vomiting, vertigo and numbness were much less frequent in flupirtine versus pentazocine recipients, and sweating, anxiety and tremor were only observed in the pentazocine group.[25]
Higher plasma concentrations of flupirtine have been observed in patients with impaired biliary function (in particular those with primary biliary cirrhosis).[53] As there is some evidence from small studies to suggest that higher plasma concentrations of flupirtine may be associated with ataxia, flupirtine is contraindicated in patients with severely impaired biliary function like primary biliary cirrhosis, liver cirrhosis and a history of hepatic encephalopathy.[53] Patients with cirrhosis (particularly those with a history of encephalopathy) may be more susceptible to developing encephalopathy and therefore caution is advised in these patients.[53] However, liver function tests indicated that flupirtine did not appear to cause further deterioration in liver function in these patients.[53] Nevertheless, as flupirtine is hepatically metabolized, the manufacturer’s prescribing information advises caution when prescribing flupirtine in patients with impaired liver function and/or alcoholism.[17]
In the clinical studies discussed, only a very small number of patients who had received flupirtine experienced abnormal elevations in liver enzymes/bilirubin during treatment in two studies: a 1-week study (n = 3[20]) and a long-term study (n = 16[47]). Values returned to within the normal range after the end of treatment in the 1-week study,[20] and after long-term treatment in patients for whom follow-up data were available.[47] No patients discontinued or had to be withdrawn from treatment because of abnormal liver function tests. The manufacturer’s prescribing information recommends monitoring of liver function at regular intervals if treatment is continued for more than 4 weeks.[17]
Although no in vitro or clinical drug-drug interaction studies with carbamazepine have been published, the potential of carbamazepine to produce hepatic enzyme induction in some patients makes the co-administration of these drugs inadvisable.[17]
In certain countries, the manufacturer’s prescribing information does not recommend the combination of flupirtine with paracetamol,[17] as in rare cases this combination led to an increase in hepatic enzymes, which decreased again on continued therapy. However, a randomized, placeboand active-controlled, double-blind, double-dummy, multiple-dose, parallel-group study in 80 healthy adult subjects did not show signs of clinically significant alterations in liver enzymes after 14-day, repeat-dose combination treatment with flupirtine (100 mg three times daily) and paracetamol (500 mg three times daily).[54]
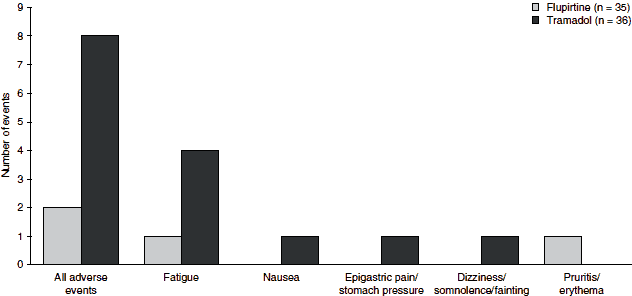 |
| Fig. 3. Adverse events in a randomized, double-blind comparison of flupirtine and tramadol for the treatment of cancer pain in outpatients (n = 71).[19] |
9. Conclusions
Flupirtine is a non-opioid analgesic with unique pharmacological properties that allow for its use in a number of disease states. Its antinociceptive, muscle-relaxant and anti-apoptotic activities without the burden of associated adverse effects seen with most opioids makes it a useful compound for the management of pain, conditions associated with increased (painful) muscle tension or spasms and a candidate to treat certain neurodegenerative diseases. It appears that flupirtine is the first representative of a unique new class of drugs denoted as SNEPCO that acts specifically via a subfamily of particular K+ channels, and this drug class is associated with a variety of potential therapeutic benefits in the treatment of pain and other disease states. Flupirtine enhances the generation of the M-current, which is a slow hyperpolarizing conductance of K+ ions that prevents the generation of neuronal action potentials and controls neuronal excitability. Flupirtine limits neuronal excitability and appears effective in the treatment of disease states that have a neuronal hyperexcitability component.
While the evidence for clinical efficacy of flupirtine mainly comes from small-scale clinical trials and the well established use in various countries over more than 2 decades, these data are promising and suggest flupirtine may have a unique and important place in pain management. However, further study in well defined clinical settings is warranted to further establish the role of this unique and fascinating agent in the management of pain and also to further delineate the full potential of its combinational use with other analgesics. Despite the limitations of the available clinical data, a series of smaller clinical studies has consistently indicated that flupirtine effectively reduces headache, and musculoskeletal, neurogenic, cancer, trauma and postoperative pain. The analgesic and muscle-relaxant properties of flupirtine are comparable to tramadol and chlormezanone, respectively, in the treatment of lower back pain. In addition, flupirtine, either alone or in combination with opioid medication, effectively reduces cancer-associated pain without an increase in sedation. When provided as combination therapy with morphine, flupirtine increases the antinociceptive activity of morphine 4-fold.
Cytoprotective, anti-apoptotic and antioxidant properties have also been associated with flupirtine use in a small number of studies to date. Flupirtine therapy displays superior tolerability when compared with commonly used synthetic opioids such as tramadol or pentazocine and does not cause tolerance or dependence.
In conclusion, flupirtine is an agent with a distinctive mechanism of action, exerting a dual therapeutic effect – with both analgesic and muscle-relaxant properties – that has utility in the treatment of pain, including that associated with muscle tension. Furthermore, preclinical data suggest that flupirtine may have neuroprotective properties. This agent, which is available in certain European countries, is currently approved for the management of pain associated with muscle tension, postoperative and post-traumatic pain, dental and cancer pain, pain associated with degenerative joint disease and trauma, headache and dysmenorrhea.
Acknowledgements
Medical writing assistance for the preparation of this article was provided by Raelene Simpson of InScience Communications, aWoltersKluwer business; technical assistance for revision of the manuscript after peer review was provided by Tracy Harrison of InScience Communications. This assistance was funded by Meda Pharmaceuticals. The author has no other conflicts of interest that are directly relevant to this review.
References



